
Caso clínico
Varón de 14 años, sin antecedentes personales y familiares de interés que presentó fiebre de hasta 40 °C axilar y odinofagia de 4 días de evolución. Fue tratado con paracetamol y amoxicilina, cediendo la odinofagia pero persistiendo la fiebre, y apareciendo decaimiento, cefalea frontal, vómitos y dolor articular. En la exploración física a su ingreso destacaba temperatura: 39,8 °C; FC 130 lat./min, FR: 35 resp./min; PA: < 85/40 mmHg; regular estado general, decaído, palidez mucocutánea intensa, frialdad distal de extremidades e hiperemia faríngea sin exudados; siendo normal el resto de la exploración.
Exploraciones complementarias
Hemograma: 22.800 leucocitos (95 % granulocitos); Hb: 14,5 g/dl; plaquetas: 208.000. Bioquímica: glucosa: 95 mg/dl. Urea: 53 mg/dl. Creatinina: 1 mg/dl. Triglicéridos: 139 mg/dl. Calcio: 9,5 mg/dl. Proteínas totales: 6,8 mg/dl. Albúmina: 3,4 g/dl, GOT: 49 U/l. GPT: 11 U/l. Ácido láctico: 0,8 mM/l. PCR: 43,5 mg/dl; coagulación: INR: 1,32; tiempo de tromboplastina activado: 30,8 s (control: 29,6 s); fibrinógeno: 712 mg/dl. LCR 0 células/μl bioquímica normal, Gram: no se observan gérmenes; sedimento de orina, ecografía abdominal y radiografía de tórax normales. Ante la sospecha de shock séptico secundario a probable foco orofaríngeo se ingresa en la UCIP realizándose tratamiento con expansión de volumen, dopamina y cefotaxima, con rápida mejoría, siendo dado de alta a planta a las 48 h. El hemocultivo y cultivo del LCR fueron negativos.
En los días siguientes volvió a presentar fiebre hasta 39 °C, acompañada de abdominalgia, hepatoesplenomegalia, exantema pruriginoso maculopapuloso confluente en tronco y extremidades fundamentalmente, y adenopatías laterocervicales 2 por 2 cm, no adheridas a planos profundos. En la radiografía de tórax se observó un infiltrado retrocardíaco izquierdo por lo que se sustituyó la cefotaxima por imipenem y se añade vancomicina. Las exploraciones complementarias realizadas dieron los siguientes resultados: serología virus de Epstein-Barr, citomegalovirus, Chlamydia pneumoniae, parvovirus B19, rubéola, herpes, toxoplasma, rosa de Bengala, borrelia, rickettsia, fiebre botonosa, leishmania, Chlamydia psitacii: negativas. Urocultivo y coprocultivos negativos. Anticuerpos antinucleares, factor reumatoide y C3 y C4 normales. Ecocardiograma, aspirado de médula ósea y gammagrafía ósea: normales; TC abdominal: hepatoesplenomegalia. Posteriormente apareció diarrea líquida, labios secos, lengua agrietada, bradicardia sinusal y taquipnea. Ante la persistencia de la fiebre con negatividad de todos los cultivos y el deterioro progresivo se retiraron los antibióticos y se inició tratamiento con ácido acetilsalicílico, gammaglobulina intravenosa y corticoterapia sin mejoría. A los 15 días de evolución presentó crisis tónico-clónicas generalizadas, precisando ingreso en UCIP y tratamiento con midazolam y fenobarbital. En la TC y la RM cerebral no se encontraron alteraciones y en el EEG se observó un foco temporooccipital izquierdo. En la analítica al ingreso en la UCIP destacaba Hb: 9 g/dl; leucocitos 3.700 (65 % granulocitos), plaquetas 107.000, fibrinógeno 82 mg/dl; triglicéridos: 926 mg/dl; ferritina 54623 μg/dl, GOT: 331 U/l y GPT: 216 U/l.
Preguntas
1. ¿Cuál es su diagnóstico?
2. ¿Con qué cuadros plantearía el diagnóstico diferencial?
3. ¿Cómo confirmaría el diagnóstico?
Síndrome hemofagocítico
Ante la presencia de fiebre prolongada, hepatoesplenomegalia, alteración multisistémica, pancitopenia progresiva y triglicéridos elevados con cultivos negativos se sospechó un síndrome hemofagocítico (SHF), aunque el aspirado de médula ósea era negativo. Se realizaron biopsia de médula ósea, hígado y piel, encontrándose en la médula ósea disminución de precursores eritroides y varias imágenes de hemofagocitosis. La fiebre persistió a pesar del tratamiento con corticoides por lo que se asoció VP16 con lo que se consiguió una rápida desaparición de la fiebre y normalización de las alteraciones analíticas.
El SHF es una entidad clínica poco frecuente 1 debida a una disfunción de la inmunidad celular, bien por una alteración funcional de las células NK (natural killer), o por una proliferación excesiva de macrófagos activados secundaria a una reacción inflamatoria exagerada. Existen dos formas: el SHF familiar (de carácter autosómico recesivo por una mutación en el gen que codifica las perforinas) 2; y el SHF secundario o reactivo a procesos neoplásicos, enfermedades autoinmunes o infecciosas (virus, parásitos o bacterias).
El diagnóstico diferencial debe realizarse con todas aquellas entidades infecciosas, neoplásicas y autoinmunes que producen fiebre y esplenomegalia importante. Nuestro paciente empezó con un cuadro compatible con un shock séptico, que mejoró transitoriamente y durante su evolución se plantearon los diagnósticos diferenciales de enfermedad del colágeno, infección por virus de Epstein-Barr, enfermedades linfoproliferativas o enfermedad de Kawasaki (por lo que recibió tratamiento con aspirina y gammaglobulina, aunque no cumpliera los criterios diagnósticos).
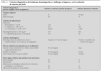
La tabla 1 presenta los criterios diagnósticos 3 internacionales, comparando con los que presentaba nuestro paciente. Pueden existir otros hallazgos como hepatomegalia, adenopatías, exantema cutáneo, alteraciones neurológicas (convulsiones, meningismo, alteración de conciencia), y prolongación del tiempo de protrombina. En ocasiones, tal como ocurrió en nuestro paciente, no se encuentran signos de hemofagocitosis en los primeros aspirados medulares. Por ello, si existe sospecha diagnóstica, es importante realizar estudios seriados de médula ósea, bazo, hígado ganglios linfáticos para poder confirmar el diagnóstico 3.

El tratamiento del SHF se realiza con corticoides intravenosos. Si no se observa mejoría deben asociarse otros fármacos como el VP16, con el que en nuestro paciente se observó una rápida mejoría 4. Otras medidas alternativas son el trasplante de medula ósea, las inmunoglobulinas a altas dosis, plasmaféresis y exanguinotransfusión 5.
En los casos familiares con tratamiento quimioterápico tan sólo se logran remisiones parciales por lo que el tratamiento de elección es el trasplante alogénico de medula ósea 6.
En los SHF secundarios la evolución es imprevisible y depende de la causa subyacente. En nuestro paciente la falta de antecedentes familiares y personales y la buena evolución tras la instauración del tratamiento orientan a un SHF reactivo o secundario, probablemente a una infección, aunque no se haya logrado establecer la etiología.
En conclusión, ante un niño con fiebre, esplenomegalia y pancitopenia progresiva de causa desconocida debe pensarse en la posibilidad del SHF. Algunos datos como la hipertrigliceridemia y la hipofibrinogenemia pueden orientar el diagnóstico, que se confirma con biopsias seriadas de médula ósea en busca de la hemofagocitosis. Ante la sospecha clínica es importante instaurar el tratamiento precozmente dado el riesgo de una evolución rápidamente fatal.
Correspondencia: Dra. M.ªJ. Santiago Lozano.
Unidad de Cuidados Intensivos Pediátricos.
Hospital General Universitario Gregorio Marañón.
O'Donnell, 50. 28009 Madrid. España.
Correo electrónico: macosantiago77@yahoo.es
Recibido en abril de 2006.
Aceptado para su publicación en mayo de 2006.
