




La obesidad es hoy la enfermedad crónica más prevalente en la infancia y la adolescencia en nuestro medio y en todo el mundo occidental. Esto la ha convertido en uno de los motivos de consulta más frecuentes en la práctica clínica pediátrica general y, particularmente, en endocrinología pediátrica. Asimismo, existe un gran número de comorbilidades secundarias a la obesidad que, cada vez con mayor frecuencia, se pueden observar ya en la infancia y en la adolescencia. Actualmente, se acepta que este gran incremento de prevalencia es debido, fundamentalmente, al desequilibrio entre la ingesta y el gasto energético propio del estilo de vida occidental. Sin embargo, cada vez es más evidente la influencia de la carga genética individual y familiar en el riesgo de desarrollar obesidad. Asimismo, se van descubriendo las bases fisiopatológicas del control del apetito y del gasto energético, a partir del estudio del creciente número de casos derivados de la existencia de alteraciones genéticas, endocrinológicas o sindrómicas subyacentes, por lo que no se puede hablar de «obesidad» de forma genérica, sino que sería más adecuado referirse a «obesidades», pues su base fisiopatológica es completamente diferente y, por tanto, difieren tangencialmente en la metodología de estudio y abordaje terapéutico.
En el año 2011, ante el niño afecto de obesidad, el pediatra debe conocer y dirigir su anamnesis y exploración hacia los signos y síntomas propios de estas entidades y, al mismo tiempo conocer los métodos disponibles en la actualidad para el diagnóstico de éstos y los recursos terapéuticos disponibles.
Obesity, as in every western country, is currently the most prevalent chronic disease in childhood in Spain. This has led to obesity being one of the most common consultations in general paediatrics and, particularly, in paediatric endocrinology. Furthermore, obesity associated comorbidities are increasing in prevalence in children and adolescents. It is widely accepted that this increase in the prevalence of obesity is derived from an imbalance between energy intake and expenditure, associated to the lifestyle in western countries. However, there is increasing evidence of the role of individual and familial genetic background in the risk of developing obesity. The pathophysiological basis of the mechanisms responsible for the control of appetite and energy expenditure are being discovered on the basis of the increasing known cases of human monogenic, syndromic and endocrine obesity. Thus it is no longer appropiate to talk about obesity but rather about «obesities», as their pathophysiological bases differ and they require different diagnostic and management approaches.
In 2011, the paediatrician must be aware of this issue and focus the clinical history and physical examination towards these specific clinical sign and symptoms, to better manage the available diagnostic and therapeutic resources when faced with a child with obesity.
La obesidad infantil constituye un motivo grave de preocupación entre los profesionales sanitarios, debido al incremento de su prevalencia observado en nuestro medio en los dos últimos decenios. Asimismo, el impacto económico actual y futuro atribuible a la obesidad en las etapas iniciales de la vida y a sus comorbilidades asociadas ha acaparado la atención de los profesionales sanitarios y de la sociedad en general, constituyéndose esta enfermedad en un tema de comentario, opinión y debate habitual en foros plurales.
Sin embargo, gran parte de estos comentarios y foros de discusión adolecen, en gran medida, del rigor necesario y exigible al referirse a esta entidad nosológica. Esto es debido, al menos en parte, a la extraordinaria complejidad que entraña el conjunto de patologías diferentes que comprende la expresión «obesidad infantil» y, por supuesto, al conocimiento aún limitado que tenemos respecto a sus causas últimas y determinantes de la acumulación patológica de tejido adiposo compartida por todas ellas.
Pese a ello, en los últimos años hemos asistido al desarrollo de múltiples líneas de investigación que nos han permitido profundizar en el conocimiento de los mecanismos reguladores del balance energético, de las bases genéticas sobre las que se produce, o no, la acumulación patológica de tejido adiposo, así como en el análisis de la función, o la disfunción, endocrinológica de éste.
En este trabajo se revisan los aspectos epidemiológicos, taxonómicos, clínicos y diagnóstico-terapéuticos actuales en obesidad infanto-juvenil, haciendo hincapié en las nuevas áreas de conocimiento desarrolladas en los últimos años.
Concepto/definiciónEl concepto intuitivo de obesidad es la acumulación excesiva de tejido adiposo que conduce a un incremento en el riesgo presente y futuro de presentar patologías asociadas, así como de la mortalidad. Este concepto, aparentemente sencillo, entraña una mayor complejidad en el periodo infanto-juvenil, debido a que, el retraso en la aparición de las comorbilidades hasta la vida adulta dificulta una definición precisa de la obesidad en función de riesgos futuros. Sin embargo la instauración de esta condición en edades tempranas permite inferir que sus consecuencias futuras puedan ser aún más graves.
La cuantificación del contenido graso corporal del niño, necesaria para la definición de obesidad, puede ser realizada de forma directa y precisa mediante técnicas específicas (bioimpedanciometría, densitometría de absorción dual de rayos X [DEXA] o hidrodensitometría)1. Sin embargo, su limitada disponibilidad, duración y coste económico han hecho que, desde la perspectiva clínica, se universalice la estimación indirecta del contenido graso corporal mediante el empleo del índice de masa corporal (IMC) o índice de Quetelet (IMC=peso [kg]/(talla [m])2). Aunque su formulación original estaba referida a individuos adultos y durante el periodo de desarrollo se proponía una modificación de la misma (kg2/m5) para referirse a la población pediátrica, se extendió internacionalmente el empleo de la fórmula original para todos los rangos etarios2.
El IMC muestra buena correlación con el contenido graso tanto en adultos como en niños3,4, si bien su interpretación en términos de contenido graso corporal experimenta variaciones de acuerdo con el sexo, la edad, el grado de maduración en niños y adolescentes, siendo sus mayores limitaciones su incapacidad para discernir el grado de desarrollo de masa muscular y para informar respecto a la distribución del contenido graso entre los distintos depósitos corporales5. Pese a estas limitaciones, el empleo del IMC como estimación indirecta del contenido graso corporal es universal y, consecuentemente, la definición de obesidad, tanto en el adulto como en el niño, se ha formulado en relación a este índice.
Así, diversos organismos internacionales, entre los que se cuenta la Organización Mundial de la Salud (OMS), avalan el establecimiento de sendos límites de IMC absoluto 25 y 30kg/m2 para definir el sobrepeso y la obesidad, respectivamente, en el paciente adulto6. Sin embargo, las diferencias en la composición corporal determinadas por la edad, el sexo y el grado de maduración puberal en el niño y adolescente hacen necesario el empleo de un valor estandarizado de IMC en función de la edad y el sexo del niño respecto de unas referencias poblacionales. Esto abre un punto de intensa controversia referente al establecimiento de los «puntos de corte» y de las referencias poblacionales que se deben emplear que, aún a día de hoy, no goza de consenso internacional.
El Grupo Internacional de Trabajo sobre Obesidad (International Obesity Task Force [IOTF]) define actualmente el sobrepeso en niños como un IMC comprendido entre los percentiles 91 y 98, de acuerdo con las referencias de Cole et al7, y la obesidad como un percentil igual o superior al 99, por medio de una extrapolación de los valores correspondientes a 25 y 30kg/m2 en el adulto, respectivamente8.
En cambio, el Centro de Control de Enfermedades de Estados Unidos de Norteamérica (Center for Disease Control [CDC]), en su informe del año 2006, formula una nueva categoría, definida como riesgo de sobrepeso, en niños cuyo IMC se sitúa entre los percentiles 85 y 94, agrupando en la denominación de sobrepeso a todos aquellos cuyo IMC iguala o supera el percentil 95 de las referencias del propio CDC del año 2000, sin referir el término obesidad9. La categoría riesgo de sobrepeso no hace referencia a un riesgo futuro, sino que indica la posibilidad de la existencia, ya en ese momento, de un exceso de grasa corporal, que precisa un estudio más específico para su confirmación.
En nuestro medio, la Guía de Práctica Clínica para la Prevención y Tratamiento de la Obesidad Infanto-juvenil10,11 postula como criterios para definir el sobrepeso y la obesidad los valores de los percentiles 90 y 97, respectivamente, específicos por edad y sexo de la distribución del IMC referido a los datos y curvas de Hernández et al12 del año 1988.
Escapa a las pretensiones de este artículo analizar en detalle la idoneidad de los umbrales y de las referencias poblacionales, más aún cuando no existen diferencias trascendentales entre ellos. Aún así, existe evidencia de que un niño presenta un exceso de grasa corporal cuando su IMC supera el percentil 95 para su edad y sexo13 y de que su definición óptima se obtiene aplicando, de forma más restrictiva, el punto de corte de +2 DE por encima del valor medio de este parámetro estimado en individuos de la misma población, edad y sexo14, coincidiendo así con la propuesta organizativa de la OMS15.
De acuerdo con lo anteriormente expuesto y, teniendo en cuenta que el establecimiento de comorbilidades asociadas a la obesidad ocurre, con frecuencia, en etapas posteriores de la vida, no es de extrañar que tampoco exista consenso actualmente sobre la definición del concepto de obesidad mórbida en la infancia y adolescencia, proponiendo algunos autores los límites de +3 SDS de IMC o 200% del peso corporal ideal para la talla como posibles «puntos de corte» para definirla.
EpidemiologíaLas dificultades para su definición y para la homogeneización internacional de los datos de referencia no impiden; sin embargo, constatar que la obesidad infantil, al igual que la del adulto, ha experimentado un incremento progresivo en los últimos 30 años en todos los países desarrollados5.
En Estados Unidos de Norteamérica se triplicó la prevalencia de sobrepeso (de acuerdo con la definición del CDC) en el rango de 2 a 19 años en este periodo; desde el 5,1% en la década de los setenta hasta el 17,1% en el registro del año 2003 a 2004. Al mismo tiempo, este fenómeno se ha visto acompañado, tanto en adultos como en niños, de un desplazamiento hacia la derecha en la curva de distribución del IMC entre los estudios epidemiológicos NHANES II (1976-1980) y NHANES 1999-2004, con un incremento más acusado de los valores más altos de la distribución6.
Múltiples estudios desarrollados en diferentes países europeos han revelado una tendencia similar, más acusada en los países de la cuenca mediterránea, con una prevalencia entre el 20 y el 40%, que en los países septentrionales5.
La prevalencia de la obesidad infantil en nuestro medio no puede precisarse debido a la ausencia de registros epidemiológicos nacionales seriados y a las diferencias metodológicas entre los estudios disponibles. Pese a todo ello, desde los datos aportados por el estudio PAIDOS ‘84, que reflejaba una prevalencia de obesidad en España del 4,9% en niños de ambos sexos16, se ha producido una progresiva tendencia ascendente, constatada por todos los estudios posteriores realizados en diferentes rangos de la edad pediátrica (estudios RICARDIN17 o PECNA18). Este último, desarrollado en la comunidad foral de Navarra, demostró un incremento en la prevalencia de obesidad desde 1987 a 1993 del 5%, siendo las cifras de 9,7 y el 14,7 en varones y mujeres, respectivamente. Posteriormente, el estudio enKid, desarrollado de forma multicéntrica entre los años 1998 y 2000 en 3534 individuos con edades comprendidas entre los 2 y los 24 años, arroja cifras de prevalencia de obesidad infantil del 13,9, el 12 y el 15,6% en niñas y niños, respectivamente, así como del 12,4% referentes a sobrepeso19. Asimismo, el estudio AVENA (Alimentación y Valoración del Estado Nutricional en Adolescentes), desarrollado en el período 2000-2002 sobre una muestra de 2.320 adolescentes con edades comprendidas entre los 13 y los 18 años, demostró una prevalencia de sobrepeso más obesidad en adolescentes de 13 a 19 años del 25,69 y 19,13% en varones y mujeres, respectivamente20.
Con posterioridad, se han comunicado diversos estudios transversales procedentes de distintas autonomías, siendo los datos nacionales más actualizados publicados por el Ministerio de Sanidad en el año 2008 los correspondientes a la última Encuesta Nacional de Salud de España (año 2006)21. Estos comunican una prevalencia conjunta de sobrepeso y obesidad en población de 2 a 17 años del 27,6% (frente al 4,9% de obesidad reportado por el estudio PAIDOS en el año 1984).
Estos datos epidemiológicos, junto con el sustancial gasto sanitario atribuido a la obesidad y a sus patologías derivadas (estimado por el Ministerio de Sanidad y Consumo en el año 2004 en unos 2.500 millones de euros anuales, equivalente a un 7% del gasto sanitario nacional anual)22, han determinado la puesta en marcha de diversas iniciativas orientadas a la prevención y a la intervención terapéutica precoz en el niño afectado de obesidad, como fue la elaboración de la Guía de Práctica Clínica para prevención y el tratamiento de la obesidad infantojuvenil10,11,23. Asimismo, se han fomentado los esfuerzos investigadores dirigidos a esta patología, como fue la creación por parte del Instituto de Salud Carlos III de una red de investigación colaborativa estable, el CIBER Fisiopatología de la Obesidad y Nutrición.
Clasificación etiológica: distintas enfermedades, un mismo nombreComo referíamos con anterioridad, una de las complejidades y de las dificultades más importantes para el adecuado entendimiento de la obesidad infantil es que, bajo el denominador común de una acumulación excesiva de grasa corporal, subyacen etiologías y, por lo tanto, entidades patológicas radicalmente diferentes. Actualmente se acepta, tanto en foros científicos como de divulgación, que el gran incremento de prevalencia de la obesidad infantil es debido al desequilibrio entre la ingesta y el gasto energético propio del estilo de vida occidental. Sin embargo, existe un porcentaje de casos derivados de la existencia de alteraciones genéticas, endocrinológicas o sindrómicas subyacentes que, si bien es cuantitativamente limitado, crece de forma continuada al tiempo que lo hacen nuestros conocimientos fisiopatológicos de la obesidad infantil.
Obesidad exógena o «común»Hasta donde nuestro conocimiento, siempre limitado, nos permite saber, ésta es la más frecuente de las entidades englobadas en la obesidad infantil. En ella, la coexistencia de una nutrición hipercalórica e inadecuadamente estructurada y de unos niveles reducidos de actividad física, propios del estilo de vida occidental actual, determinan la acumulación del exceso de energía en forma de tejido adiposo, desgraciadamente influidos a su vez por el poder adquisitivo de las familias24. Sin embargo, no todos los sujetos expuestos al mismo ambiente nutricional «obesogénico» y a similares limitaciones de actividad física desarrollan obesidad o lo hacen en similar grado. Esto es debido a que estos factores «exógenos» actúan sobre una base «endógena», la información genética propia de cada individuo, lo cual explicaría, al menos en parte, la gran heredabilidad familiar de la obesidad25.
En los últimos años, los estudios de GWAS (Genome Wide Association Studies), que podríamos traducir como «estudios hologenómicos de asociación», han perseguido, mediante el estudio de extensas cohortes de sujetos afectados de distintas patologías, hallar nuevos genes, QTL (quantitative trait loci) o haplotipos que permitan una mejor identificación del riesgo individual para el desarrollo dichas enfermedades26. Este tipo de estudios, aplicados a la obesidad, ha ofrecido una ingente cantidad de información, de modo que en la última actualización del mapa genético de la obesidad publicada, el número de genes y QTL asociados a fenotipos con afectación de la adiposidad en modelos murinos alcanzaba los 244 y 408, respectivamente27. Por este motivo, este tipo más común de obesidad debería denominarse «obesidad poligénica», pues es esta base genética la que determina la susceptibilidad del paciente ante los estímulos ambientales. Más aún, las modificaciones epigenéticas; es decir, aquellas ejercidas por dichos factores ambientales sobre el genoma de un individuo, sobre todo en fases tempranas del desarrollo, parecen desempeñar una función relevante en el riesgo individual para el desarrollo de obesidad28.
En particular, estudios recientes han asociado variantes en el primer intrón del gen «asociado a masa grasa y obesidad» (FTO) con obesidad, condicionando un elevado índice de masa corporal equivalente, aproximadamente, a +0,4kg/m2 por alelo de riesgo29–31. Así, se ha comprobado que determinados polimorfismos, en particular el rs9939609, se asocian al incremento de peso, del IMC y con los niveles de leptina en niños europeos32,33. Más recientemente (Fis34), se ha demostrado que la pérdida del gen Fto en el ratón genera retraso en el crecimiento posnatal y una reducción significativa del tejido adiposo y de la masa corporal magra. Como consecuencia, estos ratones incrementan su gasto energético, a pesar de una disminución en su actividad locomotora y su relativa hiperfagia. Estos datos podrían constituir la primera demostración directa de que Fto se encuentra involucrado funcionalmente en la homeostasis energética, mediante el control del gasto de energía.
Hasta el momento, existe constancia de variantes poligénicas en, al menos, 17 regiones genómicas independientes35 y 15 nuevos loci asociados con el IMC36. La función de estos genes candidatos se encuentra asociada a regiones que sugieren una función relevante del hipotálamo en el control del peso. También se han descrito deleciones cromosómicas raras y en pacientes con obesidad grave de inicio precoz y problemas cognitivos asociados, en particular, deleciones en 16p11.237.
Por consiguiente, el desarrollo de la obesidad en la mayoría de los niños afectados tiene una etiología multifactorial, sobre una base poligénica. Esto es, dicha base poligénica tiene per se un efecto limitado sobre el fenotipo y únicamente su combinación con otras variantes predisponentes y, sobre todo, la concurrencia de factores ambientales favorecedores del desarrollo de obesidad, determinarán finalmente el desarrollo del fenotipo obeso.
Obesidad monogénicaLa obesidad de etiología monogénica se define como la que es consecuencia de la alteración de un único gen, ya sea por deficiencia, deleción o mutación. Los pacientes afectados de este tipo de obesidad constituyen una minoría respecto al total de la población infantil con obesidad, si bien todos tienen en común la presencia de una obesidad muy intensa y de inicio precoz. Las formas monogénicas de obesidad conocidas hasta la fecha se podrían sistematizar en tres grandes categorías:
Patología en genes del sistema adipocito-hipotalámico (eje leptina-melanocortina)La red específica de neuronas productoras de poopiomelanocortina (POMC) se localiza primordialmente en el núcleo arcuato del hipotálamo, integra la información aferente sobre la energía almacenada periféricamente en el tejido adiposo que ofrece la leptina producida en aquél y señaliza mediante los productos derivados de la POMC tras su fraccionamiento por acción de la proconvertasa 1 (PCSK1), principalmente la fracción alfa de la hormona estimulante de melanocitos (α-MSH). La α-MSH actúa sobre otros núcleos hipotalámicos (fundamentalmente el núcleo paraventricular) por medio de los receptores de melanocortina (MCR), cuya isoforma número 4 (MC4R) es el principal transductor de los impulsos anorexigénicos (fig. 1). Existen detalladas revisiones referentes al control hormonal del balance energético38,39. La alteración funcional en los genes implicados en este circuito de control determina la aparición de obesidad en el niño.

Representación esquemática de la integración hipotalámica de los efectos anorexigénicos de la leptina en el núcleo arcuato hipotalámico. La flecha blanca indica su efecto estimulador sobre las neuronas productoras de POMC/CART, procesando la PCSK1 a la primera para producir α-MSH. La flecha negra indica su efecto inhibidor en las neuronas productoras de NPY/AGRP. MC4R: receptor número 4 de MSH; NPY/AGRP: neuronas productoras de neuropéptido Y y del péptido relacionado con la proteína Agouti; PCSK1: convertasa de proproteínas tipo subtilisina kexina 1; POMC/CART: neuronas productoras de proopiomelaocortina y del tránscrito relacionado con cocaína y anfetamina; RLEP: receptor de leptina; α-MSH: fracción alfa de la hormona estimulante melanocítica.
La deficiencia de leptina es una anomalía infrecuente que se hereda siguiendo un patrón mendeliano autosómico recesivo como consecuencia de mutaciones en homocigosis en el gen de la leptina (LEP, 7q31.3, OMIM #164160). La primera descripción en humanos se produjo en 199740, desde entonces, se han descrito un total de 12 individuos en el mundo, existiendo experiencia de tratamiento de estos pacientes con leptina biosintética41,42.
Estos pacientes presentan un peso normal al nacer, incrementándose de forma sustancial durante los 3 primeros meses de vida, así como ausencia de desarrollo puberal o simplemente retraso puberal, como consecuencia de su hipogonadismo hipogonadotropo, señalando la importancia de la leptina en el comienzo de la pubertad43. En consecuencia, la deficiencia congénita de leptina es singular, pues puede diagnosticarse por los niveles séricos muy bajos de leptina para la masa grasa del paciente, pudiéndose tratar con éxito mediante la administración diaria de leptina. No obstante, la secuenciación directa del gen de la leptina es aún el elemento más importante del diagnóstico.
Receptor de leptina (LEPR)La primera descripción de deficiencia del receptor de leptina por mutación en homocigosis en LEPR (1p31, OMIM +601007) se realizó en tres hermanas que presentaban una obesidad muy intensa de inicio temprano, con peso normal al nacimiento, pero con una rápida ganancia antes de los 6 meses de edad. En los 3 casos se acompañaba deficiencia de hormona de crecimiento (GH), deficiencia de tirotropina (TSH) y ausencia de caracteres sexuales en la pubertad por presentar hipogonadismo hipogonadotropo. Los niveles de leptina se encontraban elevados, proporcionalmente a la masa grasa de las pacientes44. La descripción de 8 nuevos casos ha permitido la caracterización detallada clínica y molecular de las mutaciones del gen del receptor de leptina45.
ProopiomelanocortinaLa molécula de POMC es precursora de cinco proteínas biológicamente activas: ACTH, γ-MSH, α-MSH, β-MSH y β-endorfina. La deficiencia completa de POMC conduce a la insuficiencia suprarrenal en el período neonatal, por falta de síntesis de la ACTH en las células antehipofisarias. Por consiguiente, estos pacientes requieren tratamiento con corticosteroides para prevenir crisis de insuficiencia suprarrenal.
Los primeros dos pacientes con mutaciones en POMC (2p23.3, OMIM #176830) se describieron en 199846. Posteriormente, en 2003 se describió a 3 pacientes más47. Inicialmente, el rasgo fenotípico más llamativo fue su cabello pelirrojo, interpretándose este rasgo como consecuencia de la eventual ausencia de MC1R en melanocitos; sin embargo, el sexto caso descrito se comunicó en un paciente turco48, que presentaba coloración de cabello oscura. Todos ellos mostraron un peso normal al nacimiento, con ganancia ponderal rápida en los primeros 6 meses de vida.
Convertasa de proproteínas tipo subtilisina kexina 1La convertasa de proproteínas tipo subtilisina/hexina 1 (PCSK1) es una enzima encargada del procesamiento de péptidos precursores de gran tamaño. Desempeña su acción prioritaria en el hipotálamo (junto con el subtipo 2 de PCSK) fragmentando la POMC, aunque también ejerce su acción lítica sobre la proinsulina y el proglucagón pancreáticos y, recientemente, se ha sugerido que pueda desempeñar un papel en la absorción de nutrientes en el tracto intestinal. El primer caso de mutación en el gen PCSK1 (5q15-q21, OMIM #162150) se describió en 199749 y presentaba obesidad extrema de inicio en etapas muy tempranas de la infancia, acompañada de alteraciones en el metabolismo de los hidratos de carbono, hipogonadismo, hipocortisolismo y concentraciones plasmáticas elevadas de POMC y proinsulina, así como hipoinsulinemia. Con posterioridad, se han descrito dos casos más, ambos también con hiperfagia y obesidad de comienzo temprano, probablemente por procesamiento anómalo de POMC a α-MSH en las neuronas hipotalámicas50,51.
Receptor 4 de melanocortina (MC4R)En 1998 se publicaron los dos primeros casos de obesidad en humanos por mutaciones en el gen MC4R (18q22, OMIM #155541) por dos grupos independientes52,53, cursando clínicamente con gran obesidad e hiperfagia, y siendo la causa más frecuente de obesidad humana monogénica.
En comparación con las raras mutaciones autosómicas recesivas en los genes de LEP, LEPR, POMC y PCSK1, donde se han descrito en torno a 40 casos en el mundo, la prevalencia mundial de obesidad asociada a mutaciones en MC4R se estima en torno a un 2,5%54. Más recientemente, en una amplia cohorte de pacientes obesos y controles no obesos, se apreció que la prevalencia de heterocigotos con obesidad causada por mutaciones en MC4R es del 2,6% (2,83% en niños que presentaron obesidad de comienzo temprano y 2,35% en adultos que presentaron obesidad de comienzo tardío)55.
La mayoría de las mutaciones de MC4R son heterocigotas heredadas de forma dominante, si bien se han descrito casos aislados de homocigosidad o heterocigosidad compuesta con patrón de herencia autosómico recesivo y sin fenotipo en heterocigotos. En la actualidad, no existe ningún tratamiento médico apropiado para estas anomalías. No obstante, la recuperación de expresión en la superficie celular de mutantes MC4R podría tener un beneficio terapéutico, puesto que la mayoría de las mutaciones de MC4R causantes de obesidad conducen a la retención intracelular de receptores por el sistema de control de calidad celular. En este sentido, las chaperonas podrían desempeñar una función farmacológica y, en consecuencia, ser un candidato para el tratamiento de los pacientes con mutaciones en MC4R56.
Receptor gamma para sustancias proliferadoras de peroxisomas (PPARG)-subunidad número 3, músculo-específica, de la fosfatasa 1 (PPP1R3A)El PPARG (3p25.2; OMIM: 601487) es un receptor hormonal nuclear con función reguladora en la transcripción de diversos genes. Tiene tres isoformas (G1, G2 y G3) y se considera que está relacionado con la diferenciación del adipocito y la sensibilidad a la insulina, así como con la transcripción de los genes de las proteínas desacopladoras, por lo que desempeña una función relevante en el mecanismo de la termogénesis adaptativa.
En 1998 se comunicó el hallazgo de una mutación de este gen (Pro115Gly) en 4 sujetos obesos. Las búsquedas sistemáticas subsiguientes de esta mutación demostraron su carácter excepcional57. En el transcurso del año 2002 se tuvo conocimiento de una doble mutación en heterocigosis que afectaba a los genes PPARG y PPP1R3A (OMIM: 600917, 7q31.2) en todos los miembros de una familia afectados de sobrepeso u obesidad de instauración precoz e insulinorresistencia y diabetes tipo 2 (DM2)58. El conocimiento de este hecho lo convierte en el primer caso en el que se demuestra una alteración digénica como causa de obesidad.
Patología en los genes asociados con el desarrollo del hipotálamoEn los últimos años se han descrito, en relación con el desarrollo de obesidad en el ser humano, anomalías en tres genes asociados con el desarrollo del hipotálamo: SIM1, BDNF y NTRK2. Estos genes desempeñan funciones relevantes durante el desarrollo del hipotálamo, si bien los mecanismos exactos por los que sus mutaciones de asocian al desarrollo de obesidad aún se desconocen.
SIM1La primera descripción de un paciente con obesidad extrema de comienzo temprano, obesidad, aceleración en su crecimiento y gasto energético normal por mutación en el gen SIM1 (6q16.3-q21, OMIM *603128), se produjo en una niña en el año 2000. Dicha paciente no presentaba anomalías en el desarrollo ni rasgos dismórficos, ni tampoco alteraciones endocrinológicas, mostrando una translocación de novo en uno de los alelos del gen SIM1 (Ho59).
Como quiera que los ratones con una única copia del gen Sim1 presentan el mismo fenotipo que la paciente descrita y también muestran una disminución en el número de neuronas del núcleo paraventricular, imprescindibles para el balance energético y que expresan MC4R, se ha planteado la hipótesis de que sea ésta la causa de obesidad en los ratones heterocigotos para Sim1 y en los pacientes con haploinsuficiencia para SIM1. No obstante, datos recientes sugieren que SIM1 pudiera tener una función en el balance energético aún después del desarrollo hipotalámico y, específicamente, pudiera ejercer su función en la señalización de MC4R, reguladora de la ingesta60. Más recientemente, Tolson et al61 han demostrado la función de Sim1 en la regulación de la alimentación y en la formación del núcleo paraventricular y de sus proyecciones y que la obesidad hiperfágica en ratones deficientes de Sim1 puede ser debida a cambios en el sistema leptina-melanocortina-oxitocina.
En los últimos años se han descrito raras mutaciones puntuales en SIM1 asociadas con el desarrollo de obesidad pero aún se necesitan estudios funcionales para confirmar su papel en el mismo62. De forma interesante, el año pasado se comunicó la existencia de 14 nuevas mutaciones sin sentido en SIM1 asociadas con obesidad y rasgos fenotípicos de síndrome Prader-Willi-like63.
Asimismo, se han descrito deleciones intersticiales del brazo largo del cromosoma 6 (6q14.1-q15) en varios pacientes con obesidad y un fenotipo similar al del síndrome de Prader-Willi, análogas a las que se aprecian en pacientes con mutaciones en SIM1, si bien este gen no se encuentra incluido en las deleciones intersticiales previas (6q16.3), por lo que se sugiere que estas nuevas deleciones intersticiales puedan representar un nuevo síndrome de microdeleción reconocible causado por haploinsuficiencia de los genes situados en la región 6q14.1-q1564.
Factor neurotrófico derivado del cerebroEl factor neurotrófico derivado del cerebro (BDNF9 y su receptor TRKB (tropomiosina relacionada con la quinasa B), regulan la proliferación, supervivencia y diferenciación de las neuronas durante el desarrollo, así como la plasticidad neuronal en el sistema nervioso del adulto65), la memoria, la conducta y el desarrollo cognitivo. Además, BDNF interviene en el metabolismo energético y en la conducta alimentaria66.
Una deficiencia parcial de Bdnf y Trkb en modelos de ratón originan obesidad e hiperfagia. El primer caso descrito por disrupción del gen BDNF (11p13, OMIM #113505) en el ser humano, se trataba de una niña de 8 años que presentaba obesidad e hiperfagia67, mostrando una inversión paracéntrica de novo en el cromosoma 11 que alteraba el gen BDNF en uno de los puntos de rotura cromosómicos. Aunque dicha inversión podría alterar otros genes a larga distancia o en el otro punto de rotura que contribuyeran al fenotipo del paciente, la gran similitud de este paciente con el primero descrito con mutación en el gen NTRK2 (9q22.1, OMIM #600456), el gen que codifica el receptor de BDNL, TRKB68, apoya la hipótesis de que su fenotipo era causado por haploinsuficiencia de BDNF. Asimismo, los pacientes afectados del síndrome WAGRO (OMIM #612469), causado por deleciones heterocigotas en 11p13, presentan un fenotipo que incluye obesidad y parece debido a haploinsuficiencia del gen BDNF69.
La expresión de BDNF está regulada por la señalización de MC4R en el hipotálamo ventromedial, donde se une a su receptor. Es de interés señalar que la infusión cerebral de BDNF corrige la hiperfagia en los ratones deficientes de MC4R por lo que, al igual que se comentó para SIM1, la vía de señalización de MC4R podría estar implicada en el mecanismo de génesis de obesidad en los casos de mutaciones en BDNF/NTRK2.
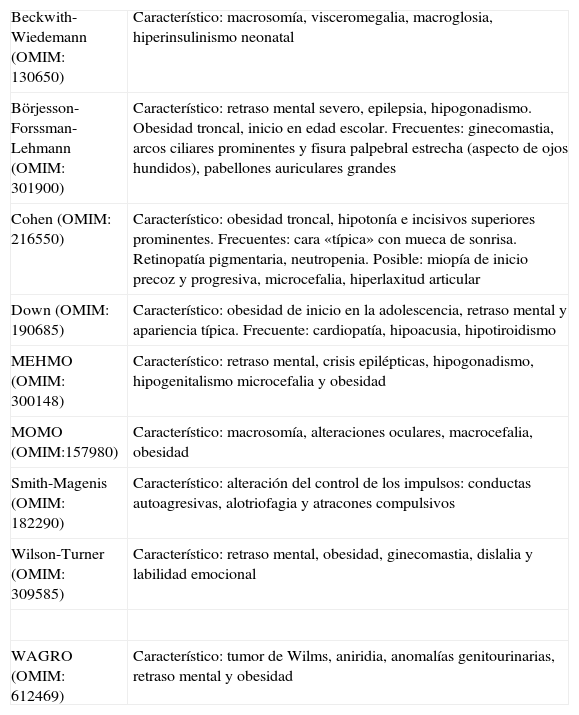
Obesidad asociada a síndromes polimalformativosSon muchos los síndromes que se transmiten con un patrón de herencia mendeliano, y que cursan con obesidad como uno de sus rasgos fenotípicos. El análisis detallado de todos ellos excede las pretensiones de esta revisión, por lo que en la tabla 1 se esquematizan las características fundamentales de aquellos que, dentro de su infrecuencia, presentan mayor prevalencia, desarrollándose brevemente a continuación los síndromes de Prader-Willi y Bardet-Biedl, en los que la obesidad constituye uno de los rasgos más destacados, con una mención a los síndromes de Alström y Carpenter, debido a los avances recientes en el conocimiento de sus bases moleculares.
Descripción clínica de los síndromes polimalformativos más comunes que presentan obesidad entre sus rasgos más característicos
| Beckwith-Wiedemann (OMIM: 130650) | Característico: macrosomía, visceromegalia, macroglosia, hiperinsulinismo neonatal |
| Börjesson-Forssman-Lehmann (OMIM: 301900) | Característico: retraso mental severo, epilepsia, hipogonadismo. Obesidad troncal, inicio en edad escolar. Frecuentes: ginecomastia, arcos ciliares prominentes y fisura palpebral estrecha (aspecto de ojos hundidos), pabellones auriculares grandes |
| Cohen (OMIM: 216550) | Característico: obesidad troncal, hipotonía e incisivos superiores prominentes. Frecuentes: cara «típica» con mueca de sonrisa. Retinopatía pigmentaria, neutropenia. Posible: miopía de inicio precoz y progresiva, microcefalia, hiperlaxitud articular |
| Down (OMIM: 190685) | Característico: obesidad de inicio en la adolescencia, retraso mental y apariencia típica. Frecuente: cardiopatía, hipoacusia, hipotiroidismo |
| MEHMO (OMIM: 300148) | Característico: retraso mental, crisis epilépticas, hipogonadismo, hipogenitalismo microcefalia y obesidad |
| MOMO (OMIM:157980) | Característico: macrosomía, alteraciones oculares, macrocefalia, obesidad |
| Smith-Magenis (OMIM: 182290) | Característico: alteración del control de los impulsos: conductas autoagresivas, alotriofagia y atracones compulsivos |
| Wilson-Turner (OMIM: 309585) | Característico: retraso mental, obesidad, ginecomastia, dislalia y labilidad emocional |
| WAGRO (OMIM: 612469) | Característico: tumor de Wilms, aniridia, anomalías genitourinarias, retraso mental y obesidad |
DM2: diabetes mellitus tipo 2; GH: hormona de crecimiento; OMIM: On-line Mendelian Inheritance in Man Database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim).
De cualquier modo, debido a las limitaciones intelectuales y físicas que presentan la mayor parte de los pacientes afectados de estos síndromes, así como debido a los tratamientos farmacológicos que reciben; el desarrollo de obesidad en estos pacientes puede deberse, en gran medida, a factores ambientales. Sin embargo, algunos de ellos, como los afectados del síndrome de Prader-Willi (PW), el PW-like, el síndrome de Bardet-Biedl o el seudohipoparatiroidismo 1A, acompañan alteraciones hipotalámicas determinantes de su hiperfagia y, por consiguiente, de su obesidad70.
Síndrome de Prader-WilliLa hipotonía neonatal y la dificultad para la succión con el subsiguiente fallo de medro son sus rasgos neonatales más característicos. La dificultad en la alimentación mejora habitualmente hacia los 6 meses de edad y, desde los 12 a los 18 meses, se desarrolla una hiperfagia incontrolable, apreciándose una disminución de la velocidad de crecimiento en la mayoría de los lactantes. La subsiguiente obesidad causada por intensa hiperfagia, unida al retraso mental, hipotonía muscular, el hipogonadismo y la acromicria, son sus características clínicas más relevantes en el periodo infanto-juvenil71.
Se trata de un cuadro clínico debido a la falta de expresión de copias paternas de genes improntados en la región 15q11-q13, fundamentalmente el gen SNRPN (small nuclear ribonucleoprotein polypeptide N) (OMIM #182279), pero también el gen NDN (necdin) (OMIM #602117) y, posiblemente, otros genes en la región pueden contribuir al fenotipo. Puede estar causado por deleciones en el cromosoma paterno, disomía uniparental materna (las dos copias del cromosoma 15 de origen materno) o por mutaciones que afectan a la impronta de la región. Aunque la región cromosómica implicada conocida ha sido extensamente estudiada72 [OMIM #176270] y se han demostrado, en contraposición a lo observado en la obesidad poligénica, niveles anormalmente elevados del péptido orexigénico gástrico ghrelina73, aún se desconoce con exactitud el mecanismo por el que se ocasiona la obesidad en estos pacientes.
Síndrome de Bardet-Biedl (OMIM 209900)Clínicamente, el síndrome de Bardet-Biedl se caracteriza por la existencia constante de retraso mental (más acusado para habilidades verbales que manipulativas) y alteraciones digitales (sobre todo polidactilia postaxial, braquidactilia o sindactilia) hasta en el 69% de los individuos en algunas series. Es frecuente la presencia de distrofia retiniana (que no es la clásica retinitis pigmentaria inicialmente descrita), anomalías renales (tanto funcionales como anatómicas [dilatación y quistes piélicos]) de inicio al final de la primera década de la vida, o microgenitalismo en varones (con niveles normales de gonadotrofinas). Es característica del subtipo 2 la presencia de colobomas iridianos74.
En estos pacientes, el incremento progresivo de masa corporal suele acontecer en torno a los 2 o 3 años de vida, si bien el subtipo 4, asociado a mutaciones en el cromosoma 15, conlleva una obesidad más intensa y de inicio precoz; siendo ésta menos intensa en el subtipo 2.
Es preciso destacar las diferencias del síndrome de Bardet-Biedl con otra entidad, el síndrome de Laurence-Moon (OMIM: 245800), con el que ha compartido la denominación de síndrome de Lawrence-Moon-Bardet-Biedl desde 1925, tras la definición formulada por Solis, Cohen y Weiss, hasta que en 1970 Ammann apuntó cómo estos pacientes diferían por su ausencia de alteraciones digitales, la menor entidad de la obesidad acompañante y la presencia, en todos ellos, de paraplejía espástica (ausente en el síndrome de Bardet Biedl), si bien el retraso mental y la retinitis pigmentaria eran rasgos prácticamente constantes75.
Del mismo modo, se hace necesario señalar la independencia nosológica del síndrome de Biemond II (OMIM: 210350), que se caracteriza por retraso mental, obesidad, hipogenitalismo, polidactilia postaxial y coloboma de iris. Este síndrome presenta un patrón de herencia de tipo autosómico dominante, con penetrancia irregular, y comparte todos los rasgos fenotípicos con el subtipo 2 del síndrome de Bardet-Biedl, por lo que ambas entidades difieren solamente en su modo de herencia76. Hasta la fecha, no se ha comunicado ningún gen asociado a los síndromes de Laurence-Moon ni Biemond II.
El síndrome de Bardet-Biedl es genéticamente heterogéneo, habiéndose descrito hasta el momento mutaciones en 15 genes distintos (BBS1-15) en relación con el mismo74,77. Aunque tradicionalmente se ha considerado una anomalía de herencia autosómica recesiva, recientemente se ha demostrado que determinados subtipos de este síndrome presentan una mutación en uno de los loci identificados para los tipos 2 a 6, junto con otra mutación en un segundo locus, lo que le ha valido el apelativo de «herencia trialélica» y, más recientemente, «herencia recesiva con modificador de penetrancia».
Al menos ocho de los genes codifican proteínas necesarias para la función ciliar neuronal primaria. Dichas proteínas forman un complejo, denominado «BBSome» que se asocia con el factor Rab8GPT para facilitar el transporte de proteínas al cilium primario74,77. Recientemente, se ha demostrado que la deleción de los cilios de las neuronas del sistema nervioso central y, más específicamente, de las neuronas que expresan POMC, produce obesidad en el ratón78. Este hallazgo pudiera ser realmente relevante, porque se trataría de la primera evidencia que explicaría el hecho de que la patogénesis de la disfunción ciliar primaria en el hipotálamo podría estar en relación con la regulación de la ingesta, siendo la causa del desarrollo de la obesidad en los pacientes afectos de síndrome de Bardet-Biedl.
Síndrome de AlströmSe trata de un síndrome heredado de manera autosómica recesiva debido a mutaciones en el gen ALMS1 (2p13, OMIM #203800). Comparte algunos hallazgos clínicos con el síndrome de Bardet-Biedl: obesidad de comienzo temprano, degeneración retiniana, DM2 y pérdida de audición; sin embargo, no presentan retraso mental, polidactilia ni hipogonadismo. Li et al79 han demostrado que ALMS1 desempeña una función relevante en la formación ciliar en las células renales.
Síndrome de CarpenterTambién denominado acrocefalopolisindactilia tipo II, cursa con craneosinostosis, polidactilia, sindactilia de tejidos blandos y obesidad. Dicho síndrome se hereda según un patrón autosómico recesivo debido a mutaciones en homocigosis en el gen RAB23 (6p11, OMIM #201000). Rab23 procede de la familia Rab de pequeñas GTPasas que regulan el tráfico intracelular de membrana asociado a proteínas. Rab23 regula de forma negativa la vía de señalización intracelular del gen Sonic hedgehog. Recientemente, Yoshimura et al80 han demostrado que Rab23 es una de las tres Rab GTPasas (Rab8a, Rab17 y Rab23) relacionadas con la formación del cilio primario.
Obesidad secundariaIndependientemente del sustrato genético individual y del balance entre ingesta y gasto energético, la presencia de obesidad en el niño puede ser consecuencia de distintas enfermedades, entre las que destacan las patologías endocrinológicas, los procesos patológicos o procedimientos terapéuticos que afectan al área hipotálamo-hipofisaria y los tratamientos farmacológicos, especialmente con principios psicoactivos. Las causas más frecuentes de obesidad secundaria y sus características esenciales se detallan en la tabla 2.
Descripción clínica de las principales enfermedades endocrinológicas y causas iatrogénicas de obesidad
| Hipotiroidismo | Defecto de producción o acción de hormonas tiroideas. Obesidad, desaceleración del crecimiento, retraso puberal y de la edad ósea, piel seca, intolerancia al frío, estreñimiento |
| Hipercortisolismo | Exceso de producción de cortisol. Desaceleración del crecimiento, osteoporosis, obesidad troncal («giba de búfalo»), estrías cutáneas, cara pletórica «de luna llena». HTA. alteración del metabolismo de los HC |
| Seudohipoparatiroidismo (osteodistrofia hereditaria de Albright) | Resistencia a la acción de la PTH. Obesidad acentuada por el hipocrecimiento acompañante. Osteoporosis generalizada, retraso en la erupción dentaria y alteraciones en el esmalte. Retraso mental en grado variable, eventualmente, hipotiroidismo o hipogonadismo |
| Hiperinsulinismo neonatal | Hipoglucemia asociada. |
| Deficiencia de GH | Hipocrecimiento posnatal severo de inicio precoz. Episodios de hipoglucemia neonatal. Acumulación de grasa subcutánea de predominio troncal. Cara redondeada, frente prominente, raíz nasal aplanada, voz aguda |
| Obesidad hipotalámica | Tumores, cirugía o radiación en área hipotálamo-hipofisaria. Característico: ausencia de sensación de saciedad e hiperfagia compulsiva. Mecanismos: pérdida de los centros reguladores de la saciedad; activación defectiva del sistema nervioso simpático (menor consumo energético y volición para la actividad física) |
| Obesidad iatrogénica | Tratamiento con: antiinflamatorios esteroideos, antidepresivos tricíclicos, fármacos neurolépticos (especialmente risperidona), ácido valproico, ciproheptadina, fármacos antihistamínicos, insulina, análogos de GnRH, hidrazidas |
GH: hormona de crecimiento; GnRH: péptido liberador de gonadotrofinas; HC: hidratos de carbono; HTA: hipertensión arterial; PTH: hormona paratiroidea; TRH: péptido liberador de tireotropina.
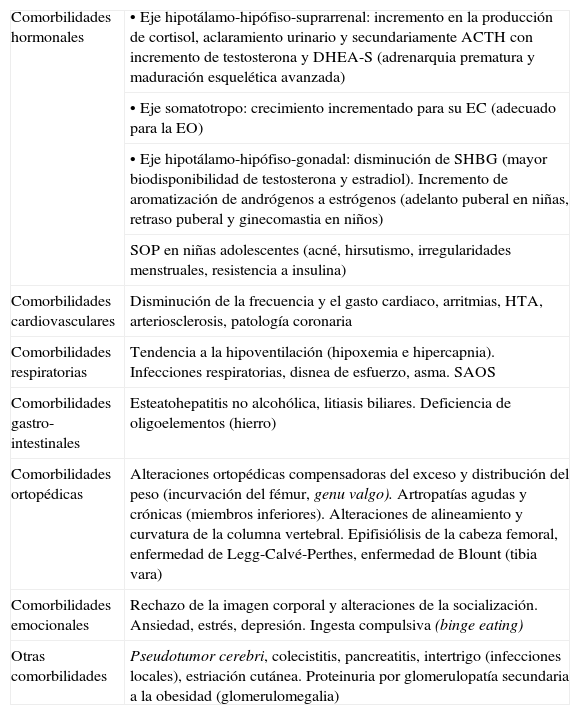
Como consecuencia del exceso de tejido adiposo es posible objetivar toda una serie de alteraciones en los diferentes órganos y sistemas, hacia cuyos signos y síntomas es preciso orientar la anamnesis, la exploración física y las eventuales exploraciones complementarias necesarias. Un análisis exhaustivo de las mismas excede nuestras pretensiones, si bien, las comorbilidades metabólicas merecen especial atención, debido a su eventual papel en el riesgo cardiovascular en la vida adulta. En la tabla 3 se enumeran las comorbilidades más frecuentemente observadas, organizadas por órganos y aparatos, remitiendo al lector para más información a nuestra revisión previa81, donde se detallan las mismas.
Comorbilidades asociadas a la obesidad y síntomas más característicos
| Comorbilidades hormonales | • Eje hipotálamo-hipófiso-suprarrenal: incremento en la producción de cortisol, aclaramiento urinario y secundariamente ACTH con incremento de testosterona y DHEA-S (adrenarquia prematura y maduración esquelética avanzada) |
| • Eje somatotropo: crecimiento incrementado para su EC (adecuado para la EO) | |
| • Eje hipotálamo-hipófiso-gonadal: disminución de SHBG (mayor biodisponibilidad de testosterona y estradiol). Incremento de aromatización de andrógenos a estrógenos (adelanto puberal en niñas, retraso puberal y ginecomastia en niños) | |
| SOP en niñas adolescentes (acné, hirsutismo, irregularidades menstruales, resistencia a insulina) | |
| Comorbilidades cardiovasculares | Disminución de la frecuencia y el gasto cardiaco, arritmias, HTA, arteriosclerosis, patología coronaria |
| Comorbilidades respiratorias | Tendencia a la hipoventilación (hipoxemia e hipercapnia). Infecciones respiratorias, disnea de esfuerzo, asma. SAOS |
| Comorbilidades gastro-intestinales | Esteatohepatitis no alcohólica, litiasis biliares. Deficiencia de oligoelementos (hierro) |
| Comorbilidades ortopédicas | Alteraciones ortopédicas compensadoras del exceso y distribución del peso (incurvación del fémur, genu valgo). Artropatías agudas y crónicas (miembros inferiores). Alteraciones de alineamiento y curvatura de la columna vertebral. Epifisiólisis de la cabeza femoral, enfermedad de Legg-Calvé-Perthes, enfermedad de Blount (tibia vara) |
| Comorbilidades emocionales | Rechazo de la imagen corporal y alteraciones de la socialización. Ansiedad, estrés, depresión. Ingesta compulsiva (binge eating) |
| Otras comorbilidades | Pseudotumor cerebri, colecistitis, pancreatitis, intertrigo (infecciones locales), estriación cutánea. Proteinuria por glomerulopatía secundaria a la obesidad (glomerulomegalia) |
ACTH: hormona corticotropa; DHEA-S: sulfato de dehidroepiandrosterona; EC: edad cronológica; EO: edad ósea; HC: hidratos de carbono; HDL: lipoproteína de alta densidad; HTA: hipertensión arterial; IHC: intolerancia a los hidratos de carbono; LDL: lipoproteína de baja densidad; RI: resistencia a la captación de glucosa inducida por insulina; SAOS: síndrome de apnea obstructiva del sueño; SHBG: proteína transportadora de esteroides sexuales; SOP: síndrome de ovario poliquístico; VLDL: lipoproteína de muy baja densidad.
La complicación metabólica más importante es la resistencia a la captación de glucosa inducida por insulina o resistencia insulínica (RI). La RI se define como la incapacidad de la insulina plasmática para, en concentraciones habituales, promover la captación periférica de glucosa, suprimir la gluconeogénesis hepática e inhibir la producción de lipoproteínas de muy baja densidad (VLDL), lo que ocasiona un aumento compensador de la secreción de insulina que puede derivar en una intolerancia a los hidratos de carbono e, incluso, en una DM2 cuando esta capacidad compensadora fracasa82.
La RI se considera la base fisiopatológica de «una serie de variables relacionadas que tienden a coexistir en el mismo individuo y que pueden ser de enorme importancia en la génesis de la enfermedad coronaria, que incluyen alteraciones del metabolismo de los hidratos de carbono, de los lípidos e hipertensión arterial», conformando lo que definió Gerald Reaven en el año 1988 como el síndrome X83. Posteriormente, se han propuesto diferentes nombres para su denominación, de los cuales, el que se estableció con más firmeza fue el de síndrome metabólico (SM)84,85, cuya importancia radica en que ayuda a identificar individuos con riesgo de desarrollar DM2 y enfermedad cardiovascular (ECV), principales causas de mortalidad en los países desarrollados. En la formulación inicial del concepto y los componentes del síndrome X por Reaven, se postulaba que el estado fisiopatológico subyacente a todos estos cambios es la RI, lo cual genera un marco fisiopatológico sobre el que poder relacionar una serie de fenómenos biológicos aparentemente no emparentados.
Esta fue la base sobre la que el grupo para el estudio de la diabetes de la OMS estableció sus criterios para el diagnóstico del SM, exigiendo, como criterio central e imprescindible la existencia de intolerancia a los hidratos de carbono (IHC), RI o DM2, junto con alteraciones en, al menos, dos del resto de componentes considerados: adiposidad (en términos de IMC o de índice cintura/cadera [ICC]), hipertensión arterial (HTA), alteraciones del perfil lipídico (reducción de lipoproteínas de alta densidad [HDL] o incremento de triglicéridos [TG]) o microalbuminuria86.
Posteriormente, estos criterios fueron revisados y reformulados tras el tercer informe del Panel de Tratamiento de Adultos del Programa Nacional de Educación para el Colesterol (NCEP-ATPIII), equiparando los criterios en importancia (sólo exige que se cumplan tres de ellos), prescindiendo de la existencia o no de microalbuminuria y evaluando la alteración del metabolismo hidrocarbonado mediante la cifra de glucemia en ayunas y la adiposidad mediante el ICC87.
Más tarde, la Federación Internacional de Diabetes (IDF) postuló una modificación sobre los criterios ATP-III en los que el cambio sustancial lo constituía el requerimiento, como condición sine quam non, de una circunferencia de cintura superior a las referencias proporcionadas para cada grupo étnico, como medida de obesidad central, junto con el cumplimiento de otros dos de los criterios88.
En el caso de los niños, el diagnóstico del SM se complica aún más puesto que, además de las diferentes definiciones existentes, por ser el niño un organismo en evolución, no son aplicables los valores absolutos utilizados para establecer los límites patológicos de cada parámetro en el adulto. Por ello y, pese a los intentos de establecer puntos de corte percentilados y ajustados a cada rango etario, algunos autores dudan de la fiabilidad de este diagnóstico89 y del verdadero beneficio de intentar aplicar este concepto en esta franja etaria, abogando por identificar y tratar estos factores de riesgo de forma individualizada90.
Ya agrupados como SM, ya considerados individualmente, se ha comprobado la asociación de todas estas alteraciones metabólicas con la obesidad y, particularmente, con la acumulación de grasa abdominal tanto en niños como en adolescentes91, con mayor frecuencia cuanto mayor es el exceso de grasa corporal92. Esto concuerda con el hallazgo de que la formación de placas de ateroma arteriales comienza ya durante la infancia y, si bien las manifestaciones de ECV no aparecen hasta la tercera o cuarta décadas de la vida, estos niños obesos ya presentan un riesgo incrementado de enfermedad coronaria cardiaca93.
En la actualidad, para establecer el diagnóstico de SM la IDF propone evaluar la presencia de obesidad troncal (perímetro de cintura) junto con las alteraciones de metabolismo de hidratos de carbono (HC) y lípidos (triglicéridos y fracción HDL de colesterol < 40mg/dl) e HTA sistólica y/o diastólica. Sin embargo, la IDF recomienda que el diagnóstico de SM no se establezca en niños menores de 10 años de edad94.
En íntima relación con el estudio de las comorbilidades metabólicas asociadas a la obesidad, se ha desarrollado en los últimos años el conocimiento de la secreción de péptidos, por parte del aparato digestivo y del tejido adiposo, encargados del ingreso y almacenaje de nutrientes, respectivamente, con implicación trascendental en aquéllas. En este sentido, ha sido fundamental la aceptación de que el tejido adiposo no es un órgano meramente pasivo, sino que expresa receptores para la mayor parte de hormonas hipofisarias e hipotalámicas y de neurotransmisores y, a su vez, se comunica mediante la secreción de numerosos péptidos y hormonas, denominados adipocinas95.
Entre ellas, se ha desarrollado el estudio de la evolución desde el periodo neonatal y a lo largo de toda la infancia del péptido orexigénico gástrico ghrelina96,97 y de la hormona anorexigénica leptina, producida en el tejido adiposo98, así como del efecto que ejerce la obesidad infantil sobre los niveles de los mismos4,99–101. Más recientemente, estos estudios se han dirigido hacia las moléculas potencialmente favorecedoras de la sensibilidad a insulina, como la adiponectina y su isoforma de alto peso molecular4, o más recientemente la visfatina o la vaspina102.
DiagnósticoComo hemos mencionado con anterioridad, las técnicas de medición directa del contenido graso corporal tienen una accesibilidad limitada en la práctica clínica cotidiana, empleándose fundamentalmente en la investigación aplicada. No obstante, la impedanciometría y, en menor medida, la DEXA y la pletismografía por desplazamiento de aire son empleadas con este fin en servicios especializados1.
Además de la cuantificación absoluta del contenido graso corporal, es fisiopatológicamente importante evaluar su distribución, ya que se conoce el papel diferencial desempeñado por los compartimentos adiposo y visceral en la eventual génesis de complicaciones metabólicas103. Para este fin, de nuevo, la estimación indirecta del contenido graso visceral realizada mediante la medición de los perímetros de cintura y cadera es el método más accesible en la práctica clínica, existiendo referencias internacionales clasificadas por grupos étnicos104. Con mayor precisión para este fin, pero al tiempo con menor accesibilidad en la práctica pediátrica cotidiana, los métodos directos de cuantificación de la grasa visceral están representados por la tomografía computarizada y, más recientemente, por la resonancia magnética105.
Una vez establecida la presencia de obesidad, si bien la mayor parte de los casos de obesidad infantil y juvenil son de base poligénica y secundarios a la existencia de un desequilibrio entre el ingreso y el consumo energético, hemos de estar atentos a los rasgos característicos de aquellos casos secundarios a mutaciones monogénicas (principalmente del receptor de melanocortina número 4 [MC4R]), enfermedades endocrinológicas o tratamientos específicos (tabla 2), puesto que las características clínicas de estos síndromes o patologías subyacentes pueden estar presentes y deben ser escrutadas en el niño o adolescente con obesidad.
Así, la anamnesis incidirá en aquellos antecedentes tanto familiares como personales que nos puedan orientar respecto a la etiología de la obesidad que presenta el paciente, haciendo especial hincapié en los hábitos nutricionales y de actividad física del niño y de la unidad familiar (tabla 4).
Aspectos importantes que se deben reseñar en la anamnesis del niño o adolescente afecto de obesidad
| Antecedentes familiares | Etnia y país de origen. Existencia de consanguinidad |
| Posibles enfermedades familiares (endocrinológicas, autoinmunitarios), haciendo hincapié en la presencia de obesidad de inicio precoz, DM2, dislipidemia, HTA o patología coronaria precoz en otros miembros de la familia | |
| Fórmula gestacional materna (gestaciones, abortos [causas], hijos vivos) | |
| IMC e hitos del desarrollo puberal de padres y hermanos (edad de telarquia y menarquia en mujeres y de inicio del incremento del volumen testicular en varones) | |
| Alteraciones menstruales, hirsutismo o SOP en mujeres | |
| Ambiente socio-económico y dinámica familiar, costumbres dietéticas (comida conjunta cotidiana, frecuencia de comidas fuera de casa, preferencias nutricionales, condicionamiento económico de la dieta) y hábitos o ausencia de ocio activo | |
| Antecedentes personales | Incidencias durante la gestación (diabetes gestacional asociada a macrosomía neonatal); fecha de primera percepción de movimientos fetales (PMF tardíos y débiles y polihidramnios asociados a síndrome de Prader-Willi) |
| Tono neonatal o necesidad de reanimación (hipotonía neonatal evidente en síndrome de Prader-Willi o Down) | |
| Edad gestacional, longitud, peso y perímetro cefálico al nacimiento (constatación de macrosomía neonatal o de niños nacidos PEG, con riesgo de obesidad y alteraciones metabólicas futuras) | |
| Hipoglucemia o ictericia neonatal (hipotiroidismo, deficiencia de GH) | |
| Tipo de lactancia y duración (defecto de succión en síndrome de Prader-Willi), pauta de introducción de la alimentación complementaria (composición de la misma y edad en el momento de introducción de cada alimento) | |
| Hitos del desarrollo psicomotor y rendimiento escolar (evaluar la presencia de retraso mental) | |
| Enfermedades y tratamientos médicos previos o actuales (tabla 2) | |
| Edad de inicio y pauta de progresión de la dentición y la pubertad (si procede) | |
| Presencia de oligomenorrea o alteraciones del ciclo menstrual en adolescentes | |
| Características de la ganancia ponderal | Edad o momento de inicio de la ganancia ponderal (muy precoz en obesidad de base monogénica) |
| Patrón evolutivo de la ganancia ponderal (instauración brusca en tumores o lesión hipotalámica) | |
| Existencia de posibles tratamientos médicos o condiciones desencadenantes (enfermedades, adenoidectomia u otras intervenciones quirúrgicas, acontecimientos adversos o estresantes) | |
| Evolución de la ganancia ponderal, aparición de síntomas o patologías asociadas e influencia de la obesidad en el comportamiento del niño | |
| Hábitos nutricionales y de actividad física | Hábitos diarios del niño referentes a duración y calidad del sueño (presencia de ronquido o pausas de apnea en el SAOS) |
| Actividad física (o juego activo en el caso de niños pequeños), actividades sedentarias (televisión, videojuegos, Internet) | |
| Encuesta nutricional (registro pormenorizado de la ingesta efectuada en las 24 o 72 h previas a la consulta) detallando, además de la composición cuantitativa y cualitativa de la ingesta | |
| La estructuración de las comidas (entorno, tiempo disponible para las Comidas, televisión o posibles distracciones acompañantes) | |
| La distribución de las mismas a lo largo del día (omisión del desayuno) | |
| La presencia o no de ingesta compulsiva (gran rapidez, necesidad de segundas porciones) | |
| El número de comidas o bebidas fuera de las comidas principales y su Composición (bebidas o «snacks» con alto contenido en HC purificados y ácidos grasos saturados y trans) |
DM2: diabetes mellitus tipo 2; GH: hormona de crecimiento; HC: hidratos de carbono; HTA: hipertensión arterial; PEG: pequeño para su edad gestacional; SAOS: síndrome de apnea obstructiva del sueño; SOP: síndrome de ovario poliquístico.
Junto a esta anamnesis detallada, se debe realizar una exploración pediátrica general, pero específicamente dirigida a la detección de cualquier signo que pueda orientar hacia la causa de la obesidad o a la existencia de comorbilidades asociadas. En ella hay que considerar especialmente:
- –
Aspecto y actitud general (distribución de tejido adiposo, acumulación en tronco [obesidad abdominal y giba de búfalo en hipercortisolismo], tono muscular, signos de retraso psicomotor).
- –
Coloración de piel y mucosas (ictericia, piel seca en hipotiroidismo), hiperpigmentación (exceso de hormona estimulante melanocítica [MSH] en la enfermedad de Cushing), hipopigmentación (deficiencia de POMC), acantosis nigricans (hiperpigmentación y engrosamiento cutáneo en cuello, axilas y/o ingles, asociada a resistencia a insulina [RI]). Presencia de estrías y coloración de las mismas (rojo-vinosas en hipercortisolismo). Acné y/o hirsutismo (SOP).
- –
Rasgos dismórficos faciales (implantación de cabello, pabellones auriculares y dientes, paladar ojival o hendido [síndromes polimalformativos]). Hipoplasia medio-facial, frente prominente, aplanamiento de la raíz nasal, cara «de muñeca» (deficiencia de GH). Plétora facial o cara de «luna llena» (hipercortisolismo).
- –
Anomalías en la visión (retinopatía asociada a síndromes polimalformativos) o en el campo visual (procesos expansivos hipofisarios).
- –
Signos displásicos (acortamiento de cuarto y quinto metacarpianos [seudohipoparatiroidismo]).
- –
Inspección y palpación de la glándula tiroidea (bocio posible en hipotiroidismo).
- –
Auscultación cardiaca y pulmonar (cardiopatías asociadas a síndromes polimalformativos).
- –
Presencia de hepatomegalia (esteatohepatitis no alcohólica).
- –
Estadio puberal (adelanto o retraso puberal). Presencia y caracterización de ginecomastia en varones. Posible presencia de adipomastia sin telarquia en niñas. Presencia de adrenarquia prematura.
- –
Presencia de alteraciones ortopédicas compensatorias (genu valgo, rectificación de las curvaturas y del alineamiento de la columna vertebral), alteraciones de la marcha o dolor a la movilización de la cadera (epifisiólisis de la cabeza femoral, enfermedad de Legg-Calvé-Perthes), aplanamiento del arco plantar.
- –
Registro de presión arterial (PA) (HTA secundaria a obesidad, especialmente en hipercortisolismo). Las determinaciones de PA deben ser percentiladas en referencia al sexo, edad y talla del individuo. El hallazgo de HTA franca en el niño, aún en el contexto de la obesidad, conlleva la necesidad de un estudio detallado, incluidos un registro continuado de PA, un estudio cardiológico para evaluar su posible repercusión retrógrada y un estudio nefrológico para descartar causas renales de la misma.
La Academia Americana de Pediatría, por medio de su comité de expertos, recomienda estudiar en todos los niños con obesidad los niveles de transaminasas (aspartato aminotransferasa y alanina aminotranferasa), glucemia basal y perfil lipídico106, recomendación esta última también avalada por la Asociación Americana del Corazón107. Sin embargo, se ha podido comprobar cómo la aparición de RI y ulteriores alteraciones del metabolismo de los HC y de los lípidos, es un proceso progresivo108, por lo que puede existir una hiperinsulinemia franca, en ausencia de AGA4. Por este motivo, sería aconsejable incluir la determinación de insulinemia basal en la evaluación de los niños y adolescentes obesos, lo que a su vez permitirá el cálculo del índice HOMA (glucosa [mmol/l] x insulina [μU/ml]/22,5), indicador de resistencia a insulina.
El resto de las exploraciones complementarias a realizar estarán determinadas por los datos relevantes de la anamnesis y los hallazgos de la exploración física. Así:
- –
La realización de una radiografía de mano y muñeca izquierdas (tobillo en niños menores de 2 años) permite establecer la «edad ósea» (EO) para evaluar el ritmo madurativo del paciente en relación con su talla, edad cronológica (EC) y estadio de desarrollo puberal. En la obesidad es frecuente una EO acelerada respecto a la EC (pero adecuada a la talla del niño). La EO muestra retrasos significativos respecto a la EC en el hipotiroidismo o la deficiencia de GH y aceleraciones evidentes en el hipercortisolismo y la pubertad precoz.
- –
Los exámenes bioquímicos nos permitirán la evaluación del ionograma (la natremia y la сaliemia puede verse alteradas en el hipercortisolismo), del calcio y fósforo plasmático (seudohipoparatiroidismo); urea y creatinina séricas y presencia de microalbuminuria (nefropatía). Asimismo, por la posible asociación de su deficiencia, es aconsejable estudiar el hemograma y las concentraciones de hierro y ferritina109. Actualmente, no existe consenso respecto a la necesidad de determinar sistemáticamente la uricemia en la obesidad infanto-juvenil, si bien es una práctica habitual.
- –
Aunque no existe consenso al respecto en la obesidad infantil, se debe considerar la realización de una prueba de tolerancia oral a la glucosa (TTOG) en aquellos casos en los que el paciente pertenezca a un grupo étnico de riesgo (hispano, afroamericano) y/o existan alteraciones de la glucemia (> 100mg/dl) o insulinemia basales (> 15μU/ml), dislipidemia, HTA, antecedentes familiares de DM2, o condiciones asociadas a la RI, tales como acantosis nigricans o síntomas del SOP. Por medio de estos estudios se pueden definir las siguientes entidades diagnósticas: alteración de la glucemia en ayunas (AGA, > 100mg/dl); intolerancia a los HC (IHC, glucemia tras 2h en el TTOG=140-199mg/dl), DM2 (glucemia en ayunas > 126mg/dl o tras 2 horas del TTOG > 200mg/dl, repetidas en 2 ocasiones). En este último caso, es preciso registrar las cifras de hemoglobina glucosilada, que desde el año 2010 es también propuesta por la American Diabetes Association como marcador diagnóstico de prediabetes (5,7-6,4%) o diabetes (si supera el 6,5%)110.
Pese a la escasa prevalencia de estas entidades, los estudios hormonales deben ir dirigidos a descartar la existencia de hipotiroidismo (tiroxina libre y TSH) o de hipercortisolismo (excreción urinaria de cortisol de 24h). Ante la sospecha de deficiencia o insensibilidad a GH, se debe incluir la determinación de los niveles de factor de crecimiento similar a la insulina número 1 (IGF-I) e proteína transportadora de IGF número 3 (IGFBP-3). Actualmente, se ha propuesto la posible utilidad de la determinación de los niveles de adiponectina circulante, en particular de sus multímeros de alto peso molecular, como posible indicador del riesgo de desarrollo de alteraciones en el metabolismo de los HC por su relación inversa con la RI4.
La ecografía es la prueba de elección ante la sospecha de la existencia de esteatohepatitis no alcohólica o SOP111. Por otra parte, en aquellos casos en los que se sospeche un estado hipometabólico o en los que no se observa un adecuado resultado de la restricción dietética, puede estar indicada la realización de una calorimetría indirecta para determinar el gasto energético basal del paciente y así poder ajustar con más precisión sus requerimientos nutricionales.
El estudio de las concentraciones circulantes de adipoquinas (hormonas segregadas por el tejido adiposo como leptina, receptor soluble de leptina o interleucina 6, entre otras) o de posibles mutaciones monogénicas no está indicado de forma sistemática en el estudio de la obesidad infantil, debiéndose, por el contrario, solicitar ante la sospecha clínica de estas entidades y confirmando los hallazgos por medio del estudio molecular correspondiente siempre que esté disponible.
La presencia de signos o síntomas sugerentes de comorbilidades específicas determinarán la necesidad de realizar una evaluación psicológica o de ampliar la evaluación médica especializada (digestiva, cardiológica, ortopédica, nefrológica) o los exámenes complementarios (estudio polisomnográfico en síndrome de apnea obstructiva del sueño).
Mención especial merece la agrupación de un conjunto de comorbilidades metabólicas asociadas a la obesidad bajo el concepto de SM, pese a la discusión activa existente en lo referente a la idoneidad de emplear este término en los niños. A este respecto, los criterios formulados en el año 2007 por la IDF para el diagnóstico del SM en niños y adolescentes, con la presencia de obesidad abdominal como requisito obligatorio, son los siguientes94:
- –
Edad 6 a 10 años: el SM no puede ser diagnosticado, pero hay que prestar atención individualizada a las comorbilidades presentes y a la historia familiar, recomendándose la reducción ponderal cuando el perímetro de cintura (cin) alcanza o supera el percentil 90 de las referencias por grupo étnico.
- –
Edad 10 a 16 años: cin > p90 por grupo étnico (o límite de adulto, si es menor) junto a 2 o mas de: triglicéridos >150mg/dl; HDL <40mg/dl; PA sistólica (PAS) > 130mmHg o diastólica (PAD) > 85mmHg; glucemia en ayunas >100mg/dl o DM2 diagnosticada.
- –
Edad igual o superior a 16 años: cin > 94cm para varones caucásicos y > 80cm para mujeres caucásicas, junto a 2 o más de: triglicéridos > 150mg/dl o en tratamiento específico, HDL < 40mg/dl (varones) o < 50mg/dl (mujeres) o en tratamiento específico; PAS > 130mmHg o PAD > 85mmHg o en tratamiento específico; glucemia en ayunas > 100mg/dl o DM2 diagnosticada.
Las recomendaciones actuales proponen la instauración de acciones dirigidas al mantenimiento ponderal en los niños de entre 2 y 6 años cuyo IMC supere el percentil 95 respecto a su edad y sexo. Mientras tanto, estas medidas deben tener como objetivo la reducción ponderal en los niños mayores de 6 años con IMC > p95, así como para niños de cualquier edad que padezcan complicaciones asociadas al exceso de peso106.
Por lo tanto, el abordaje terapéutico del niño afectado de obesidad debe acometerse en el momento del diagnóstico, sin demorarse hasta edades futuras hipotéticamente más adecuadas para el mismo. Más aún, la sistematización de los escasos estudios de estrategias de intervención disponibles dirigidas hacia un impacto ya en edades muy tempranas (0-5 años), apoya la idea de que los padres son receptivos y capaces de adoptar hábitos que favorezcan el desarrollo de un peso normal en sus hijos112.
Actualmente, existen múltiples guías de práctica clínica disponibles, siendo las más recientes la publicada por el Ministerio de Sanidad español10 y la de la Scottish Intercollegiate Guideline Network113. El tratamiento de la obesidad infantil se basa en tres elementos, que son: la reorganización de los hábitos alimentarios, la actividad física y el tratamiento comportamental. En cambio, el tratamiento farmacológico o quirúrgico tiene aún una indicación excepcional en el período juvenil. No obstante, las revisiones sistemáticas y los metaanálisis de la literatura disponibles respecto a las diferentes opciones terapéuticas coinciden en señalar la imposibilidad, en el momento actual, de establecer recomendaciones específicas a este respecto, o de precisar los resultados de estas intervenciones a largo plazo114,115.
Estos resultados van a verse influenciados en gran medida por el acercamiento inicial del pediatra al paciente, a su problema y al núcleo familiar. Asimismo, la naturaleza de las intervenciones que se instauren y el modelo de seguimiento que se plantee van a ser determinantes en el resultado final obtenido.
El seguimiento del niño afectado de obesidad necesariamente se extenderá durante un periodo prolongado, por lo que no debe limitarse al ámbito hospitalario, si bien existen evidencias consistentes en que este tratamiento debe ser instaurado por un profesional experimentado y que su implicación debe extenderse al seguimiento del paciente al menos durante su periodo inicial. Así, una vez establecido el vínculo inicial de confianza con el niño, el seguimiento contará con más posibilidades de éxito si es realizado, siempre que sea posible, por el mismo facultativo y con una frecuencia de visitas inicialmente más alta. Sobre esta base, existe la posibilidad de incluir a los pacientes en programas de atención individualizada, o bien en grupos terapéuticos116, de acuerdo con esta disponibilidad de tiempo del facultativo e instalaciones de la institución responsable del tratamiento. En el momento actual, no existe evidencia para recomendar una de estas modalidades de atención sobre la otra, ni una periodicidad ni duración específica de las visitas, siendo necesario, una vez más, individualizar el tratamiento en función de las características del niño y de la familia.
Tratamiento conductualEl objetivo de este tratamiento es ayudar al niño a adquirir nuevas habilidades que le permitan alcanzar unos objetivos previamente consensuados. En este abordaje cabe distinguir dos componentes fundamentales: las técnicas de modificación de conducta y la terapia dirigida al estrés.
La mayor parte de los estudios incluyen el tratamiento conductual en el contexto de un abordaje generalizado de cambios en el estilo de vida, que también contempla modificaciones en la alimentación y en la actividad física. Estos ensayos sugieren que la terapia cognitivo-conductual puede contribuir a disminuciones moderadas en el IMC en adolescentes con obesidad a corto plazo. Por este motivo, las distintas guías de práctica clínica coinciden en recomendar la inclusión de estas técnicas cognitivo-conductuales y de la terapia dirigida al estrés como parte de un programa integral de cambios en el estilo de vida, ya sea de forma individual o grupal y dirigiéndola a todo el núcleo familiar.
Técnicas de modificación de conductaSu base es el principio del condicionamiento clásico o respondiente, siendo la ingesta alimentaria la conducta evocada y los estímulos evocadores aquellos a los que el paciente asocia un aumento de ingesta (ver televisión, reuniones con amigos y celebraciones, entre otras), con una fuerza de asociación entre estímulos y conducta tanto mayor cuanto más veces se concatenan117. El objetivo será identificar y combatir aquellos estímulos (situaciones, emociones) que conducen a la pérdida de control sobre la ingesta alimentaria y/o el sedentarismo, favoreciendo la aparición de comportamientos no deseados.
Asimismo es necesario el análisis de la recompensa o refuerzo que para cada individuo se deriva de las actividades que fomentan o inhiben la ganancia de peso, bajo la premisa de que aquellas actividades que nos reportan placer, se tienden a repetir, fomentando la concesión de refuerzos positivos tras el logro de determinadas metas comportamentales previamente establecidas118.
Terapia dirigida al estrésEl estrés puede favorecer actividades no saludables, como la ingesta compulsiva de alimentos, e inhibir conductas beneficiosas, como el ejercicio físico. Estas terapias se dirigirán a la identificación y modificación de los pensamientos y sentimientos automáticos, así como de los derivados del fracaso en la obtención de los objetivos marcados. Este abordaje cognitivo permite, asimismo, establecer objetivos y evaluar los logros en los cambios de la alimentación y la conducta de forma realista119.
En el caso de los niños, los objetivos a alcanzar deben ser claros, comprensibles para ellos, alcanzables y fácilmente medibles, para reforzar la percepción subjetiva del éxito derivado de la consecución de los mismos. Además, hay que hacer hincapié en describir y ayudar a descubrir los mecanismos que se pueden emplear para lograr dichos cambios (cómo cambiar) por encima de los comportamientos susceptibles de ese cambio (qué cambiar). El elemento prioritario es la automonitorización, acompañada del control de los estímulos, la reestructuración cognitiva, la solución de los problemas derivados de su aplicación a la práctica y la prevención de recaídas119.
Como hemos mencionado anteriormente, la adecuada relación del pediatra con el paciente y un tiempo de atención suficiente permiten el desarrollo de la denominada «entrevista motivacional» orientada a alcanzar los objetivos expuestos, con resultados satisfactorios demostrados, sobre todo cuando se implica al núcleo familiar120.
Intervención nutricionalTambién las intervenciones nutricionales evaluadas para el tratamiento de la obesidad infantil se engloban, mayoritariamente, en estrategias combinadas de cambios en el estilo de vida, como antes mencionábamos en relación al tratamiento conductual. Más aún, la comparación de los tratamientos nutricionales tienen la dificultad añadida de que estos ensayos tienen, en general, baja calidad metodológica y presentan una gran variabilidad en el tipo de intervención, su duración, tiempo de seguimiento, ámbito de aplicación y grupo de comparación. Por otra parte, no se evalúa su efecto sobre la reducción ponderal a largo plazo. Así, es constante la referencia a la imposibilidad de establecer recomendaciones específicas respecto a la intervención nutricional en la obesidad infantil y la necesidad de ensayos controlados con seguimiento a largo plazo.
Estas fuentes incluyen la evaluación de dietas con distinto grado de restricción calórica, alteración de la composición de macronutrientes (limitadas en grasas, HC o ricas en fibra o proteínas) o micronutrientes (calcio).
Entre ellas se distingue un primer grupo constituido por las dietas con alteraciones en las proporciones de macronutrientes, sobre cuya efectividad y limitaciones existe literatura científica, tanto en pacientes adultos como en niños, mostrándose que las dietas bajas en carbohidratos y con índice glucémico bajo son tan efectivas como las hipocalóricas para la pérdida de peso a corto plazo121. En este grupo, recientemente se ha comunicado el resultado comparativo de dietas con bajo aporte de HC y grasas, respectivamente en adolescentes, mostrando las primeras una mayor efectividad a corto plazo, pero similares resultados a medio plazo (12 semanas)122. Sea como fuere, no existe experiencia en el tratamiento con estas dietas a largo plazo en adolescentes y precisan, sobre todo las dietas con aportes limitados de HC, una evaluación pretratamiento y un seguimiento clínico cercano, por los efectos secundarios metabólicos que pueden presentar123.
Sin embargo, existe un segundo grupo de dietas, denominadas heterodoxas, sin fundamento fisiopatológico demostrado y respecto a las cuales, o bien no existe literatura científica o bien ésta no apoya su hipotético fundamento científico124. Debido a la proliferación exponencial de estas últimas, la Agencia Española de Seguridad Alimentaria y Nutrición, dependiente del Ministerio de Sanidad y Consumo español, ha elaborado un listado de las mismas, realizando una valoración nutricional de aquellas empleadas con más asiduidad (http://www.aesan.msc.es/aesa/web/AESA.jsp).
En cualquier caso, no existen estudios sistemáticos rigurosos de la eficacia ni de la seguridad de estas dietas en el niño, por lo que no se aconseja su empleo en la edad pediátrica. Asimismo, las dietas extremadamente restrictivas en el aporte calórico o aquellas en las que se realiza una sustitución por compuestos alimenticios líquidos o purificados proteicos no son recomendables debido a la alta posibilidad de deficiencias nutricionales y complicaciones asociadas, si bien se han empleado con éxito por centros especializados en casos de adolescentes con obesidad mórbida y patología concomitante grave.
Por el contrario, las intervenciones nutricionales en la obesidad infantil deben estar dirigidas a la organización de la ingesta y a la reducción del exceso calórico en la alimentación del niño, a expensas de la utilización de alimentos con alto valor nutricional, de forma equilibrada, con una limitación moderada de la ingesta energética, de modo que puedan ser mantenidas en el tiempo y siempre en combinación con otras intervenciones conductuales y de ejercicio físico10,113. En esta línea, la organización y distribución de las comidas es el primer paso para la adecuada estructuración de los ingresos energéticos y de principios inmediatos, puesto que los alimentos ingeridos habitualmente en los periodos comprendidos entre las comidas suelen ser ricos en grasas e HC purificados, evitando periodos de ayuno prolongado. En cuanto a la ingesta de líquidos, una vez satisfechos los requerimientos lácteos diarios, la bebida habitual debe ser el agua, minimizando las bebidas azucaradas. Estas directrices deben acompañarse de dos medidas conductuales referentes a la alimentación que han mostrado su efectividad en la limitación del exceso de ingesta calórica: la reducción del tamaño de las raciones y la ralentización del acto de la ingesta, favoreciendo así la sensación de saciedad119.
Así, el comité de nutrición de la Asociación Española de Pediatría recomienda en el niño y el adolescente afectado de obesidad una alimentación mixta, variada, cuantitativamente limitada por medio de una restricción calórica moderada125. Para favorecer el aprendizaje y la aplicación de estas recomendaciones por parte del propio niño, se han desarrollado instrumentos como la «dieta del semáforo» (traffic light o stoplight diet), desarrollada por Epstein et al en los años setenta en la Universidad de Pittsburgh, basada en la categorización de los alimentos en tres grupos, de acuerdo con su aporte de grasa y asociados a un color (verde, amarillo o rojo). Así los alimentos «verdes» contienen entre 0 y 1,9g de grasa por ración, los «amarillos» entre 2,0 y 4,9g y los «rojos» 5g o más, lo que determina la frecuencia recomendable de su consumo y siendo el objetivo final promover el consumo de alimentos «verdes» y disminuir la de los «rojos»126.
Actividad físicaExiste evidencia concluyente de que los niños y adolescentes obesos desarrollan menos actividad física que sus coetáneos no obesos127, así como del efecto beneficioso de la reducción de las actividades sedentarias y del incremento de la actividad física sobre el peso, la composición corporal y las comorbilidades metabólicas asociadas a la obesidad en niños y adolescentes128, alcanzando ambas recomendaciones un alto grado de evidencia en las guías de práctica clínica disponibles.
Para alcanzar el primer objetivo se aconseja la retirada del televisor y/o el ordenador de las habitaciones de los niños y se insiste en la necesidad de limitar y programar de antemano, por parte de los padres, tanto los contenidos como el tiempo máximo dedicado a entretenimientos sedentarios. En relación con la actividad física, es necesario insistir en la necesidad de incrementar la actividad derivada de la vida cotidiana, alentando los traslados a pie y evitando los medios mecánicos, siempre que sea posible. En cuanto a la actividad física específica que se debe desarrollar, ésta debe adecuarse a la edad del paciente y resultar atractiva y, sobre todo, divertida para él, modificándose conforme éste va creciendo. Debe transmitir al niño seguridad en su desempeño, evitando la posibilidad de situaciones que sienta comprometidas o peligrosas y no conllevar, al menos inicialmente, estrictos requerimientos de intensidad. En una segunda etapa, de acuerdo con la adquisición de habilidades y la mejoría física por parte del niño, se puede incrementar gradualmente la duración y/o intensidad de la actividad, colectivizándola cuando el niño se sienta suficientemente seguro para ello129. Por este motivo, no existe consenso sobre la pauta exacta de frecuencia, duración e intensidad de las sesiones de ejercicio físico a desarrollar, debiendo ser adecuadas en función de la valoración individualizada de cada paciente y modificadas conforme este entrenamiento progrese.
Tratamientos excepcionales en la obesidad infantil:La consideración de intervenciones terapéuticas adyuvantes (fármacos, técnicas quirúrgicas u otros) a los cambios en el estilo de vida en la obesidad de la infancia y adolescencia ha surgido como consecuencia del incremento de la prevalencia de comorbilidades graves, incluso potencialmente letales, en este rango etario. Sin embargo, la cantidad y la calidad de evidencia al respecto de ellas son tan limitadas como los estudios disponibles al respecto.
Por estas razones, estas opciones se contemplan exclusivamente en situaciones particulares y graves, limitándose habitualmente a la adolescencia y en presencia de comorbilidades asociadas.
Tratamiento farmacológicoCon independencia de aquellos fármacos antaño empleados, pero carentes de indicación actual en el tratamiento en adolescentes (dietilpropión, fenfluramina), en los últimos años se han retirado dos principios activos más: el rimonabant (empleado exclusivamente en adultos) y la sibutramina (previamente aceptado por la FDA en EE. UU. para el tratamiento en adolescentes mayores de 16 años).
Así las cosas, el único principio activo actualmente disponible para el tratamiento de la obesidad es el orlistat, ya que la metformina (un fármaco sensibilizante a la acción de la insulina) no cuenta con la obesidad entre sus indicaciones terapéuticas, si bien existen revisiones sistemáticas recientes que reflejan una reducción significativa del IMC tras el empleo de la misma en pacientes obesos130,131.
El orlistat es un inhibidor de la lipasa pancreática y gástrica, que interfiere la absorción del colesterol y los ácidos grasos libres contenidos en la dieta, con una absorción limitada del propio fármaco. Existen escasos ensayos controlados del empleo de este fármaco en adolescentes, también con resultados dispares respecto a la reducción del IMC132–134, e incluso un estudio piloto en niños prepuberales135. Por este motivo, no existen recomendaciones consistentes referidas a su uso en este rango de edad, sugiriéndose únicamente que podría constituir una ayuda al tratamiento habitual en los adolescentes afectados de obesidad, al menos en los 12 primeros meses del mismo, en la reducción del IMC10, en una postura apoyada por los resultados derivados de un metaanálisis de reciente publicación136, e incidiendo en la necesidad del empleo de suplementos de vitaminas liposolubles, debido a la interferencia que ejerce el fármaco respecto a su absorción. Asimismo, se destaca que los efectos adversos gastrointestinales pueden limitar su empleo, que no existe evidencia para recomendar una dosis óptima y que se desconocen los efectos a largo plazo.
Tratamiento quirúrgicoLos datos referentes a cirugía bariátrica en niños y adolescentes son extremadamente reducidos, limitándose a comunicaciones de series de casos y a consensos de expertos, sin ensayos controlados ni datos referentes a los resultados a largo plazo. Por consiguiente, no pueden formularse recomendaciones referentes a su empleo en niños y adolescentes, limitándose su uso a casos de obesidades o comorbilidades excepcionalmente graves, realizándose en centros altamente especializados10,113.
Para considerar a un adolescente candidato a la cirugía bariátrica, los comités de expertos solicitan, además una serie de requisitos antropométricos (IMC > 40kg/m2) y la presencia de comorbilidades graves asociadas, la constatación de madurez tanto corporal (estimada mediante la madurez esquelética) como cognitiva, del paciente, así como valorar su capacidad de decisión y estructura familiar, con el fin de reducir la posibilidad de efectos adversos, tanto en la intervención como en el posterior seguimiento. Además, se establece como requisito el fracaso previo de los programas intensivos de pérdida de peso, durante un periodo mínimo de 6 meses137. Por lo tanto, los niños o adolescentes tributarios de cirugía bariátrica constituyen una mínima proporción, al igual que son escasos los centros provistos del equipo multidisciplinar y la experiencia necesarios para la realización y el seguimiento de estas intervenciones.
La serie retrospectiva de casos comunicada por el Grupo Colaborativo para el Estudio de Cirugía Bariátrica Pediátrica se basa en tratamiento mediante by-pass gástrico, técnica más utilizada en EE. UU., en la que predomina el componente restrictivo, con un trayecto intestinal prolongado de circulación conjunta de bolo alimentario y secreciones biliopancreáticas. Esta serie, que incluye a 39 adolescentes tratados, comunica una reducción significativa de IMC, insulina, TG y colesterol tras el tratamiento138. En Europa y Australia, en cambio, se ha empleado con más asiduidad la implantación de la banda gástrica ajustable, técnica restrictiva consistente en la aplicación laparoscópica de una banda de silicona que circunda la porción proximal del estómago, bajo la unión gastroesofágica, y cuyo calibre puede regularse mediante la inyección de suero salino en un reservorio subcutáneo, con resultados esperanzadores en adolescentes139.
Otros tratamientosUn procedimiento terapéutico ampliamente publicitado para el tratamiento de la obesidad es la implantación endoscópica de dispositivos (balones) intragástricos. Su fundamento radica en favorecer la sensación de saciedad. Se ha comprobado la seguridad en su implantación que, incluso, se puede realizar bajo sedación, sin necesidad de anestesia. Sin embargo, no deja de ser un procedimiento transitorio, ya que el dispositivo debe ser retirado tras 6 o 7 meses de su implantación, comunicándose un efecto beneficioso en la reducción ponderal leve, reversible y dependiente del tiempo. Asimismo, los efectos secundarios gastrointestinales (náuseas, vómitos, dolor) tras su implantación son prácticamente universales, no estando exento de complicaciones potencialmente graves como la perforación o la migración intestinal140. Las series de adolescentes comunicadas son prácticamente inexistentes, incluyen muy pocos pacientes y sus resultados son poco alentadores141.
Actualmente, se encuentran en fase de estudio y desarrollo nuevos tratamientos, que incluyen los denominados alimentos funcionales, aquellos que aportan un efecto beneficioso más allá de sus propiedades nutricionales, como el ácido linoleico conjugado, fármacos que mimetizan el efecto de las señales periféricas de saciedad o nuevos agentes con acción central, aunque sin evidencia suficiente en este momento para formular recomendaciones al respecto de su utilización.
Tratamientos en desarrolloLa alta prevalencia, no sólo en la infancia y adolescencia, sino en todos los rangos etarios, de obesidad en todo el mundo está haciendo que el esfuerzo investigador en el potencial tratamiento farmacológico de la obesidad se multiplique en los últimos años. Así, la reversión del cuadro clínico ocasionado por la deficiencia de leptina observada tras la administración de leptina recombinante en los pacientes afectados41, abre la puerta al desarrollo de nuevos fármacos cuya diana terapéutica sea el MC4R, por ser esta la causa más frecuente de obesidad monogénica en el ser humano, como hemos mencionado anteriormente. Si bien los ensayos clínicos disponibles hasta el momento son escasos142,143, con efectos modestos en la reducción ponderal y efectos secundarios moderados (rubefacción, náuseas, vómitos, cefalea, somnolencia y alteraciones del gusto).
Asimismo, están en desarrollo nuevos fármacos con mecanismo de acción tanto a nivel periférico, como a nivel central, que se han revisado exhaustivamente en recientes revisiones144–146. Se podrían agrupar según su efecto en:
- 1.
Acción periférica destinada a disminuir la absorción de nutrientes:
- –
Nuevos inhibidores de la lipasa pancreática (cetilistat).
- –
Inhibidores de la proteína microsomal intestinal de transporte de triglicéridos (MTP), sin esteatorrea como efecto secundario en los ensayos comunicados147.
- –
Inhibidores de acil-transferasas (DGAT y MGAT).
- –
- 2.
Generación de estímulos anorexigénicos e inhibición del orexigénico:
- –
Agonistas del receptor GPR119 (ODA, OEA), relacionado con la secreción de péptido similar a glucagón tipo 1 (GLP-1) y PYY.
- –
Análogos de péptidos intestinales asociados a la saciedad (fig. 2): péptido YY; oxintomodulina, colecistocinina, o péptido similar a glucagón tipo 1 (GLP-1) (exenatida y liraglutida), con indicación actual en el tratamiento de la diabetes por su efecto como incretinas.
; α-MSH: fracción alfa de la hormona estimulante melanocítica.") Figura 2.
Figura 2.Representación esquemática de la integración hipotalámica de las señales aferentes relacionadas con la homeostasis energética procedentes del sistema digestivo y del páncreas. Las flechas blancas indican efecto estimulador; la de color negro, efecto inhibidor. La ghrelina es el único estimulador de las señales orexigénicas, pues el PYY se une al receptor Y-2, presináptico, inhibiendo la vía de señalización NPY/AGRP. GLP-1, CCK y el polipéptido pancreático tienen diversos mecanismos de acción, pero todos ellos efecto anorexigénico. CCK: colecistocinina; GHSR: receptor del secretagogo de la GH; GLP1: péptido similar a glucagón tipo 1; MC4R: receptor número 4 de MSH; NPY/AGRP: neuronas productoras de neuropéptido Y y del péptido relacionado con la proteína Agouti; POMC/CART: neuronas productoras de proopiomelaocortina y del tránscrito relacionado con cocaína y anfetamina; PYY: péptido YY 3-36; RLEP: receptor de leptina; YR: receptor Y; Y2R: receptor Y subtipo 2 (presináptico inhibidor); α-MSH: fracción alfa de la hormona estimulante melanocítica.
- –
Análogos de péptidos pancreáticos asociados a la saciedad: amilina (pramlintida); polipéptido pancreático (TM-30339).
- –
Antagonistas de la acción de la ghrelina mediante bloqueadores de los receptores GHSR-1a148, estrategias de aptámeros de ARN149 o inmunización150.
- –
- 3.
Incremento periférico del gasto energético:
- –
Fármacos agonistas del receptor beta adrenérgico número 3 (CL316243) con postulado efecto lipolítico y termogénico.
- –
Fármacos miméticos de las hormonas tiroideas, con acción selectiva sobre su receptor (eprotiroma y sobretiroma).
- –
Fármacos inhibidores de la 11-bhidroxiesteroide deshidrogenasa tipo 1 (AMG331), catalizador de la transformación de cortisona (inactiva) a cortisol (activo), especialmente en tejido adiposo.
- –
Fármacos agonistas de la sirtuina 1 (SIRT1), implicada en la modulación de la lipólisis y la lipogénesis.
- –
Fármacos agonistas de TGR5 (INT-777), que ocasionan un aumento del gasto energético y de la liberación de GLP-1.
- –
- 4.
Modulación central del apetito y del gasto energético:
- –
Tratamientos con fármacos antiepilépticos (topiramato y zonisamida) y antidepresivos (bupropión), cuyos ensayos no han progresado más allá de la fase III debido a la presencia de efectos secundarios. Actualmente, se está ensayando el empleo de tesofensina, un inhibidor de la recaptación presináptica de serotonina, dopamina y noradrenalina, con resultados esperanzadores en fase II y escasos efectos secundarios.
- –
Combinaciones de fármacos (bupropión-naltresona [Contrave©], fentermina-topiramato [PHEN/TPM CR; Qnexa©], bupropión-zonisamida [Empatic©]) con formulaciones que controlan su velocidad de liberación con el objetivo de incrementar su eficacia y minimizar sus efectos secundarios151,152.
- –
Nuevos fármacos agonistas inversos del receptor de cannabinoides (Taranabant) que ocasionan un descenso del apetito y un incremento de la lipólisis y termogénesis periférica153.
- –
Factor ciliar neurotrófico y análogos sintéticos (axoquina): cuyo mecanismo de acción consiste en la supresión del gen del neuropéptido Y (NPY), principal neurotransmisor orexigénico en el hipotálamo154 y con una característica interesante, como es la ausencia de reganancia ponderal tras el cese del tratamiento155. Si bien, los ensayos no han podido progresar de la fase III por objetivarse el desarrollo, en la mayor parte de los pacientes, de anticuerpos frente al fármaco.
- –
Análogos de la hormona de crecimiento (AOD9604-AOD9401), que son los dominios de la molécula específicamente responsables de la actividad lipolítica y que permiten su administración oral en ausencia del resto de efectos metabólicos indeseables (resistencia a insulina, retención de sodio) de la molécula completa156.
- –
Antagonistas del receptor de la hormona concentradora de MCR (MCH; NGD4715), hasta ahora sólo validados en modelos animales157.
- –
Inhibidores de la proteín tirosín fosfatasa tipo 1 (PTP1B), que interfiere la correcta señalización de la leptina en el hipotálamo (MSI-1436).
- –
Antagonistas de los neurotransmisores orexigénicos hipotalámicos NPY y del péptido relacionado con la proteína Agouti (AgRP) (TTP435).
- –
Agonistas de receptores serotoninérgicos subtipo-2 (M-clorofenilpiperazina [mCPP], lorcaserina)158 y subtipo-6159.
- –
Antagonistas de receptores histaminérgicos subtipo-3 (A-331440, HPP404).
- –
Proteincinasa 2 dependiente de Ca++/calmodulina, con efecto inhibidor de la expresión de NPY hipotalámico en modelos animales160.
- –
; α-MSH: fracción alfa de la hormona estimulante melanocítica.")
En este artículo hemos intentado realizar una actualización de los aspectos taxonómicos, epidemiológicos, fisiopatológicos, clínicos, diagnósticos y terapéuticos de la obesidad en el periodo de la infancia y la adolescencia en el momento actual, en los que, como en muchas otras patologías, presenta singularidades que la diferencian de la afección en la etapa adulta. Hemos pretendido resaltar la ausencia de una definición universalmente aceptada para la misma ni para la obesidad mórbida y el incremento en su prevalencia en el momento actual, lo que la convierte en uno de los motivos de consulta más frecuentes en la práctica clínica pediátrica cotidiana.
Hemos resaltado que es preciso un cambio de la mentalidad de los pediatras en el abordaje de estos pacientes, haciéndose necesaria la consideración de distintas «obesidades» o enfermedades diferentes que convergen en un mismo rasgo fenotípico, el aumento del peso corporal. Existen signos y síntomas sugerentes de las mismas, a los que el pediatra debe prestar especial atención y que esto puede determinar diferencias en los métodos de exploración complementaria que nos puedan permitir un adecuado diagnóstico de estos pacientes. Igualmente, que la carga genética de cada niño y las modificaciones que, sobre ella, pueden ejercer los factores ambientales, sobre todo en etapas tempranas de la vida, son determinantes para su riesgo de desarrollar obesidad.
Por otra parte, hemos reseñado cómo, aún hoy en el siglo xxi, la base del tratamiento de la obesidad infantil y juvenil está constituida por el cambio en el estilo de vida que debe ser abordado en una triple vertiente (conductual, nutricional y de actividad física) dirigida al paciente y a su entorno. Finalmente, hemos intentado reflejar el estado actual de los tratamientos farmacológicos y quirúrgicos disponibles en aquellos casos en los que, durante la adolescencia, existen condiciones y comorbilidades de especial gravedad para el paciente.
Finalmente, hemos esbozado un gran número de principios activos farmacológicos, actualmente en desarrollo, que dan idea del esfuerzo investigador actual en esta área. Queda por delante, sin embargo, el reto de conseguir alcanzar tratamientos efectivos y aplicables en este rango etario y, de forma más acuciante, el reto de conseguir estrategias efectivas y eficientes para la prevención la obesidad, que permitan una aplicación poblacional universal y que conduzcan al control epidemiológico de la denominada pandemia del siglo xxi.
- –
Concepto de obesidad: no existe un acuerdo internacional. Es de utilidad en la infancia considerar un índice de masa corporal ≥ P97 para edad y sexo, con el empleo de tablas de referencia para la población a estudio.
- –
Concepto de obesidad mórbida: no hay un acuerdo internacional. El establecimiento de un índice de masa corporal > +3 DE para edad y sexo, con el empleo de tablas de referencia para la población a estudio, podría ser sensato.
- –
Concepto de obesidad de inicio temprano: no hay un acuerdo internacional. Definirla como la que se inicia antes de los 5 años de edad puede ser adecuado para enfocar estudios específicos. Otros autores, por el contrario, incluyen niños < 10 años de edad.
- –
Concepto de obesidad común: la más frecuente en la población general, que no se encuentra asociada a ninguna entidad sindrómica ni tiene base monogénica.
- –
La obesidad «común» tiene una base poligénica.
- –
Concepto de sobrepeso: no existe un acuerdo internacional. Es de utilidad en la infancia considerar un índice de masa corporal IMC ≥ P90 y < P97 para edad y sexo, con el empleo de tablas de referencia para la población a estudio.
- –
El ambiente nutricional en el feto influencia el riesgo de desarrollar obesidad en la vida adulta, afectando el desarrollo neuroendocrino del hipotálamo. Por consiguiente, la nutrición perinatal y en la lactancia temprana se cree tiene una influencia programada sobre los neurotransmisores hipotalámicos, que determinará el desarrollo de la obesidad en la época adulta. Estos hechos se apoyan en las observaciones de que tanto la obesidad como la diabetes mellitus tipo 2 pueden ser alteraciones hipotalámicas y que los nacidos con bajo peso presentan, en edades posteriores, una prevalencia elevada de obesidad, diabetes mellitus tipo 2 y síndrome metabólico.
- –
La obesidad es una enfermedad neuroendocrinológica: la demostración de enfermedades monogénicas por afectación de la expresión de ciertos genes, así como el síndrome de disfunción ciliar primario, han demostrado una función prioritaria del hipotálamo y las neuronas que conectan sus núcleos.
- –
Se conocen al menos 20 enfermedades monogénicas causantes de obesidad.
- –
La existencia de obesidad grave e hiperfagia en un niño de menos de 5 años, con historia familiar de obesidad de inicio temprano, justificaría la realización de estudios genéticos.
- –
El desarrollo de microarrays de SNP de alta densidad y su aplicación en el cartografiado de autocigosidad, proporciona un método eficiente para identificar nuevos genes relacionados con la obesidad.
- –
Los estudios de asociación del genoma completo serán imprescindibles para detectar los genes implicados y explicar la susceptibilidad individual.
Los autores declaran no tener ningún conflicto de intereses.
FinanciaciónCIBER Fisiopatología de la Obesidad y Nutrición (CB06/03). Fundación de Endocrinología y Nutrición. Instituto de Salud Carlos III, Fondo de Investigación Sanitaria (FIS: PI09/91060 y PI10/00747).
