




La sarcoidosis es una enfermedad crónica multisistémica de etiología desconocida. La histiocitosis eruptiva generalizada (HEG) es una forma rara de histiocitosis no Langerhans, de curso benigno y autorresolutivo.
Presentamos el caso de una niña de 8 años de edad con sarcoidosis que fue inicialmente diagnosticada en forma errónea de HEG. Fue tratada con corticoides orales, metotrexato y adalimumab, con los que se logró un control insuficiente de la uveítis. La introducción de infliximab permitió un control del compromiso oftalmológico y reducir la dosis de corticoides.
En algunos casos de sarcoidosis la falta de granulomas bien organizados en el inicio de la enfermedad, así como la presencia de células gigantes, puede sugerir diagnósticos alternativos como la histiocitosis no Langerhans. Aunque la experiencia del uso de antagonistas del factor de necrosis tumoral α en niños con sarcoidosis es limitada, puede ser útil en aquellos pacientes con enfermedad severa y refractaria.
Sarcoidosis is a chronic multisystemic granulomatous disease of unknown origin. Generalised eruptive histiocytosis is a rare, benign, self-healing, non-Langerhans’ cell histiocytosis (non-LCH).
We report the case of an 8-year-old girl with sarcoidosis who was misdiagnosed as non-LCH. She was treated with oral corticosteroids, methotrexate and adalimumab, but there was insufficient control of ocular disease. The introduction of infliximab achieved a control of the uveitis and enabled the corticosteroid dose to be tapered.
In some cases of sarcoidosis the lack of well-organised granuloma formation at the beginning of the disease, and the presence of prominent giant cells may suggest alternative diagnoses, such as non-LCH. Although the experience of tumour necrosis factor-α antagonists use in children with sarcoidosis is limited, these drugs may be helpful for those patients experiencing a severe and refractory disease.
La sarcoidosis es una enfermedad infrecuente en niños, caracterizada por la formación de granulomas epitelioides no caseificados, de etiología desconocida y presentación clínica variada. Frecuentemente produce infiltración pulmonar, linfadenopatías hiliares, lesiones oculares y cutáneas1,2. El diagnóstico se realiza sobre la base de la clínica y el hallazgo de granulomas epitelioides circunscritos, sin necrosis central ni corona linfocitaria3.
La histiocitosis eruptiva generalizada (HEG) es una forma infrecuente de histiocitosis, caracterizada por la erupción de múltiples pápulas amarronadas o rojoazuladas en la cara, el tronco y la región proximal de los miembros, de curso benigno y autorresolutivo4.
Existen casos límite entre estas 2 enfermedades, difíciles de diagnosticar inicialmente, en los que la falta de formación de granulomas claros en algunas lesiones cutáneas, así como la presencia de células gigantes multinucleadas prominentes tipo-Touton, pueden llevar a diagnósticos equivocados.
Los corticoides sistémicos son el tratamiento de elección de la sarcoidosis, pero existen numerosos medicamentos «ahorradores de corticoides» que han mostrado ser efectivos (principalmente el metotrexato). Hay evidencia de que el factor de necrosis tumoral alfa (TNF-α) está involucrado en la patogénesis de la sarcoidosis, por lo que los inhibidores del TNF-α se han utilizado como tratamiento. Presentamos un caso de un niña con sarcoidosis de inicio en la infancia que inicialmente fue diagnosticada de histiocitosis no LC y tratada con anti-TNF-α.
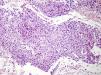
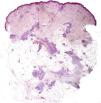
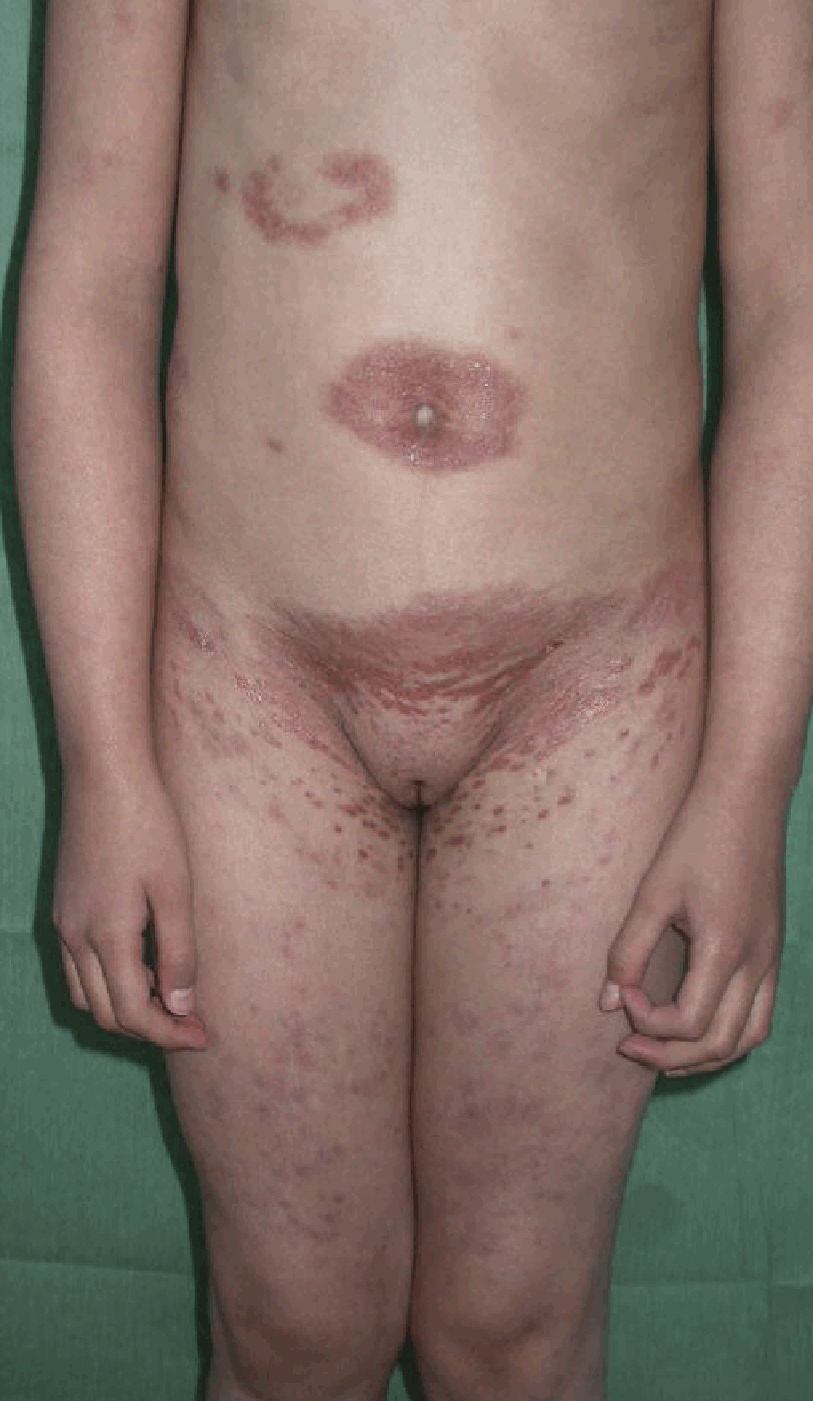
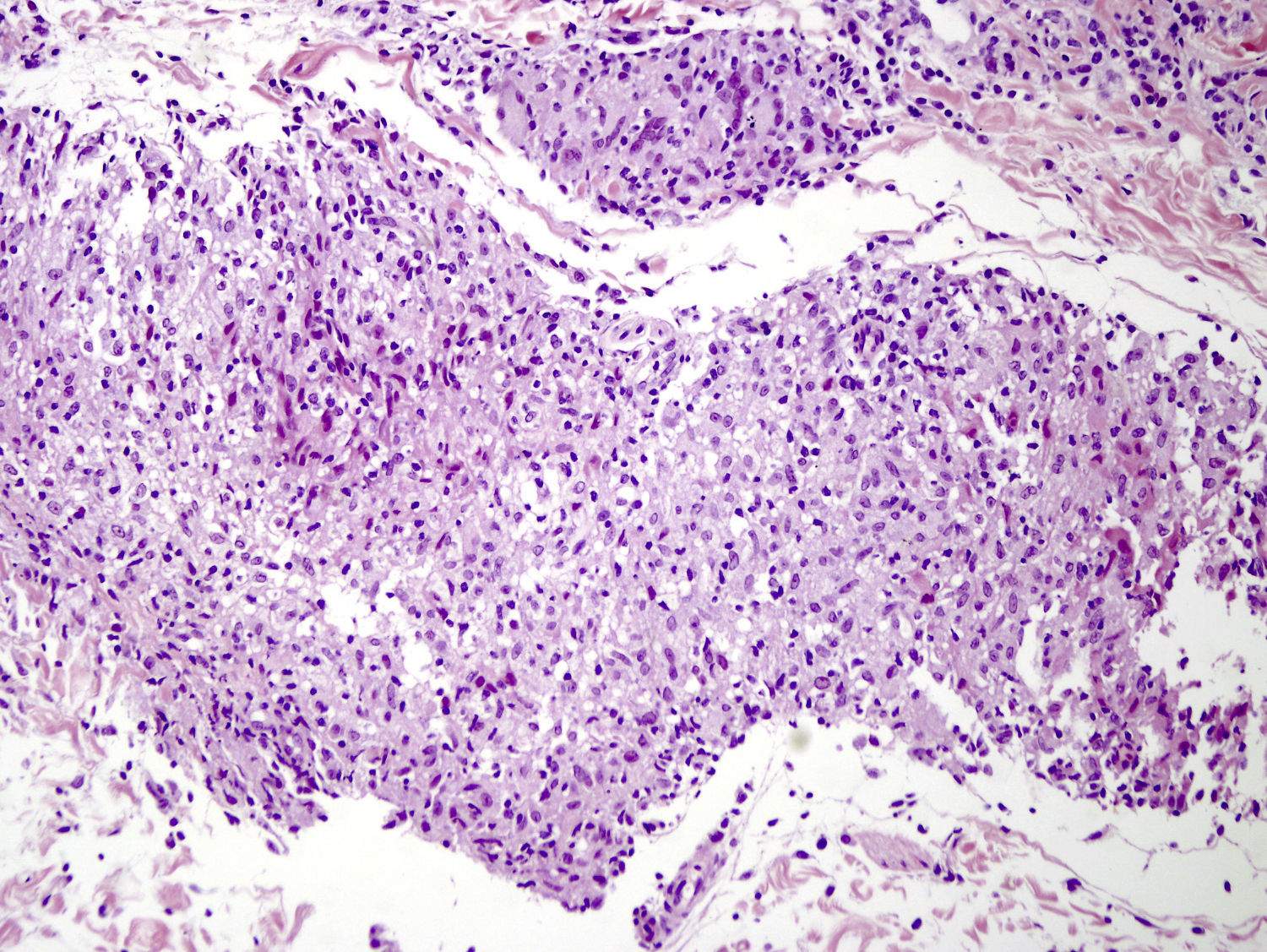
Caso clínicoSe trata de una niña de 8 años de edad, con historia de panuveítis desde los 5 años, que consultó por lesiones cutáneas de 6 meses de evolución acompañadas de artritis de codos y rodillas. Al examen físico presentaba lesiones tipo pápulas planas eritematoanaranjadas de hasta 0,5cm, algunas con tendencia confluente formando placas, infiltradas, lineales, de distribución simétrica, en la región pubiana e inguinal, circulares en región periumbilical y abdomen, afectando además la región lumbar y raíz de miembro superior derecho. En los miembros inferiores presentaba nódulos de hasta 1cm eritematovioláceos (fig. 1). Las analíticas fueron normales o negativas (hemograma, bioquímica, orina, autoinmunidad, HLAB27, PCR, calcio urinario, Mantoux y serologías frente a VHC, VHB, VIH). El estudio histopatológico mostró un denso infiltrado histiocitario localizado en la dermis reticular y tejido celular subcutáneo, constituido por histiocitos de citoplasma amplio y eosinófilo que en la zona profunda estaba xantomizado, acompañado de células gigantes multinucleadas, algunas de aspecto tipo Touton (fig. 2). La inmunohistoquímica mostró que los histiocitos eran CD68+ y S1000– y CD1a–, con lo que se diagnosticó de histiocitosis no LC, tipo HEG. Se inició metotrexato 18mg/semana y las lesiones cutáneas mejoraron dejando cicatrices deprimidas. Un año después presentó un empeoramiento de la uveítis, de la artritis y un nuevo brote de lesiones. Otra biopsia cutánea mostró resultados similares a la anterior. Se añadió adalimumab 20mg/15 días y presentó una importante mejoría. Permaneció asintomática a nivel cutáneo y articular, aunque con varias reagudizaciones del compromiso oftalmológico, por lo que se aumentó la dosis de adalimumab a 40mg/15 días y requirió ciclos de prednisona por vía oral. Dos años después presentó nuevo brote de lesiones cutáneas y una nueva biopsia mostró en la dermis reticular y el tejido celular subcutáneo un infiltrado inflamatorio linfohistiocitario (S100–) con formación de granulomas epitelioides bien definidos, sin necrosis y con células gigantes, de distribución perivascular y perianexial. PAS y Ziehl, negativos (figs. 3 y 4). Las analíticas fueron normales, a excepción de la enzima convertidora de angiotensina (ECA), 114,7 U/L (VN: 8,0–52,0 U/L), y el Mantoux negativo. No se realizó el estudio genético de la mutación de NOD2 por falta de disponibilidad del mismo en nuestro laboratorio. Una TC pulmonar mostró adenopatías paratraqueales e hiliares derecha alguna con calcio. Las pruebas funcionales respiratorias mostraron una leve restricción. Se diagnosticó de sarcoidosis de inicio en la infancia. Se aumentó la dosis de prednisona a 1mg/kg/día y se cambió a infliximab inicialmente a dosis de 6mg/kg, con aumento posterior a 15mg/kg cada 4-6 semanas (que continúa recibiendo hasta el momento en total 7 meses), presentando importante mejoría de la uveítis, desaparición de las adenopatías hiliares y de las lesiones cutáneas, con lo que se logró disminuir la dosis de prednisona.
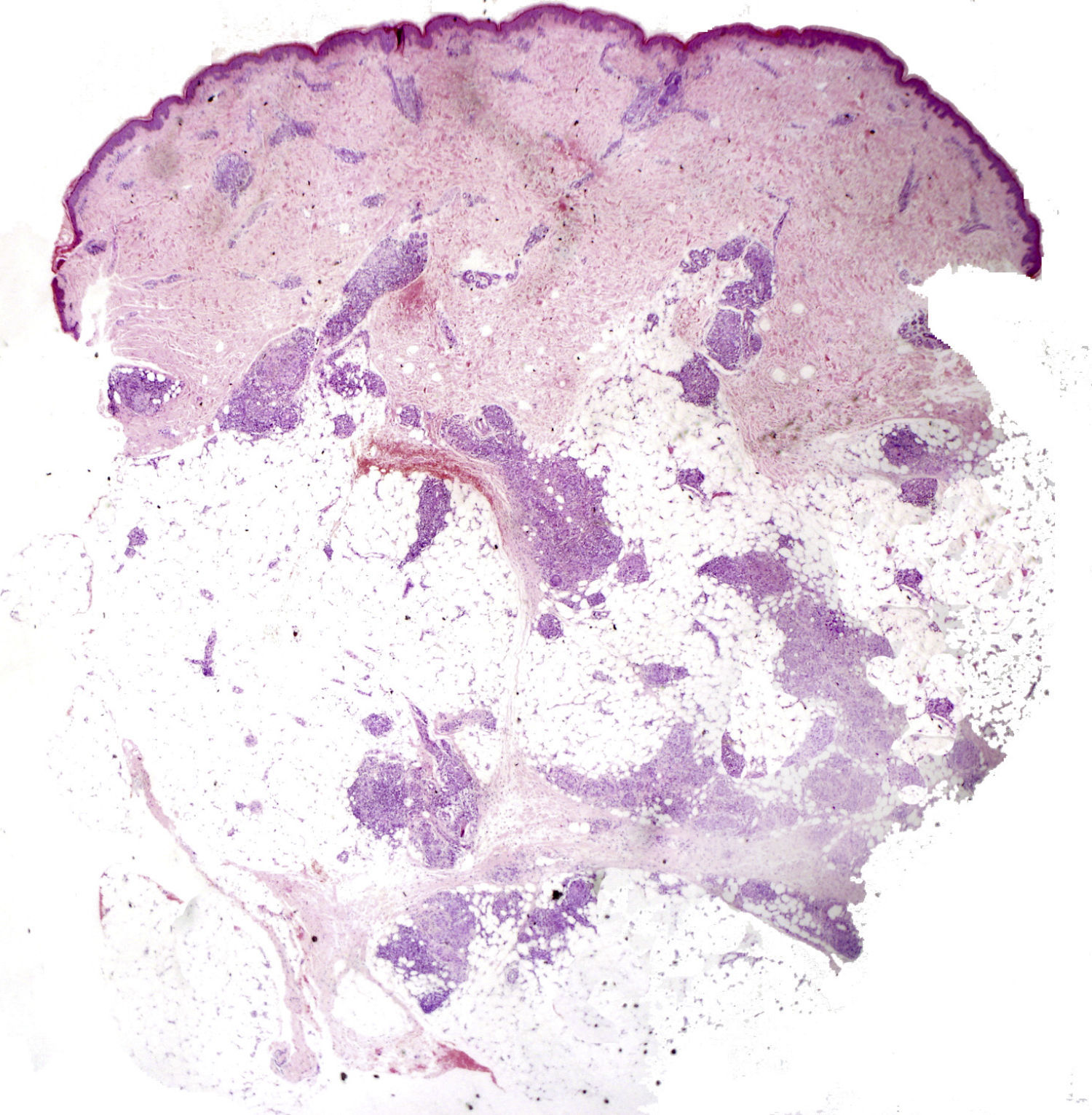
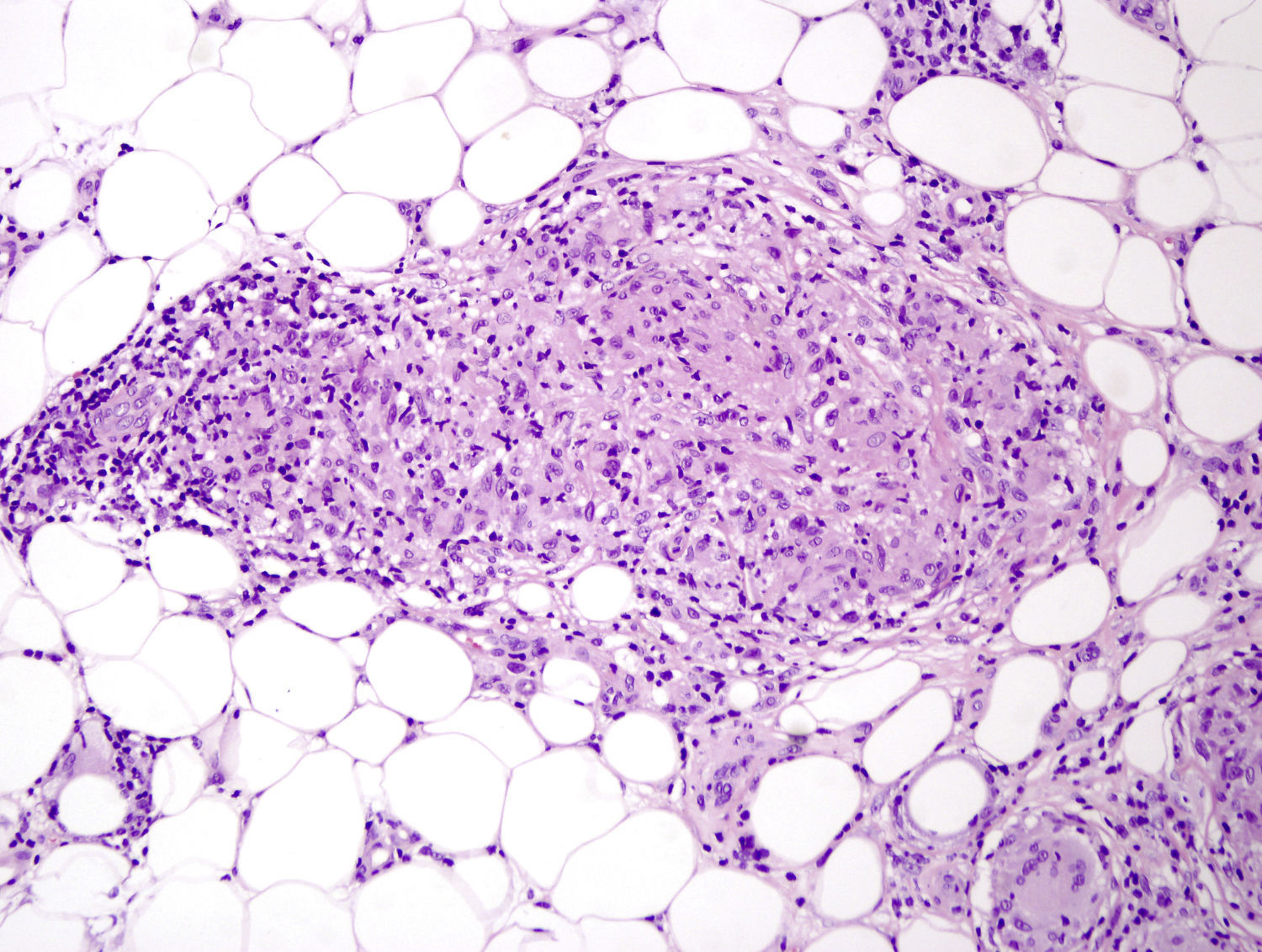
La HEG es una forma benigna de histiocitosis infrecuente, descrita inicialmente por Winkelmann y Muller en 19634. Su patogenia es desconocida; se cree que se trataría de una proliferación reactiva de histiocitos (se la ha relacionado a virus herpes 6, a neoplasias y a fiebre reumática).Se caracteriza por la erupción recurrente de múltiples pápulas, asintomáticas, de color amarronado o rojoazuladas, simétricamente distribuidas en la cara, el tronco y la región proximal de los miembros. Es autorresolutiva (o dejando maculas hiperpigmentadas). En casos típicos, histológicamente presenta una infiltración monomorfa de células mononucleares histiocíticas, en la dermis superficial y media. Los histiocitos muestran núcleos grandes con cromatina escasa y clara y citoplasma vacuolizado. Estas células muestran marcadores inmunohistoquímicos de células no LC (CD68+, CD1a–, S100–) y factor XIIIa+. Se observan, además, escasos linfocitos. La presencia de células gigantes multinucleadas es inusual pero no descarta el diagnóstico.
La sarcoidosis es una enfermedad de etiología incierta, distinguiéndose 2 formas clínicas diferentes en niños. Usualmente, en niños mayores la enfermedad se manifiesta de forma similar a la de los adultos, con frecuente afectación pulmonar, de ganglios linfáticos hiliares y ocular; mientras que la sarcoidosis de inicio temprano que afecta generalmente a niños menores de 5 años es una forma particular, que presenta típicamente la tríada de lesiones cutáneas, artritis y uveítis, siendo el compromiso de los ganglios linfáticos hiliares infrecuente5. En este caso, las primeras manifestaciones (oculares) se iniciaron a los 5 años de edad con compromiso posterior cutáneo, articular y pulmonar, comportándose como una sarcoidosis del adulto.
No está claro el papel de la ECA en el diagnóstico de sarcoidosis. Dicha enzima se encuentra elevada en el 50% de los niños con sarcoidosis de inicio tardío, pero no es específica de esta entidad6,7. Además, en niños sanos el nivel de la ECA en suero es mayor que en los adultos8.
El diagnóstico de sarcoidosis se realiza sobre la base de la presentación clínica sumada al hallazgo, en los tejidos afectados, de granulomas que centralmente tienen colecciones de macrófagos epitelioides (CD68+ y CD1a–) sin corona linfocitaria.
El diagnóstico diferencial se debe realizar con enfermedades infecciosas, neoplásicas e inflamatorias. Se deben descartar infecciones granulomatosas con tinciones especiales y cultivos, especialmente las causadas por micobacterias y hongos. En ocasiones, se debe diferenciar de enfermedades neoplásicas como los linfomas (en casos con adenopatías hiliares), con la artritis reumatoide juvenil (en casos en los que predomina el compromiso articular), con desórdenes metabólicos como el hiperparatiroidismo primario (en casos con hipercalcemia). El síndrome de Blau, descrito inicialmente en 1985, es una entidad que histológicamente es muy difícil de diferenciar de la sarcoidosis de inicio temprano; ambas, además, comparten la presencia de mutaciones genéticas en gen CARD15 que se localiza en la región pericentromérica del cromosoma 16 (16q12) y codifica para la proteína NOD2, la cual actúa como un receptor intracelular de componentes bacterianos, concretamente a peptidoglicano9,10. El síndrome de Blau se diferencia de la sarcoidosis de inicio temprano por presentar herencia autosómica dominante, inicio usualmente antes de los 4 años y afectación secuencial de la piel, las articulaciones y los ojos, sin afectación pulmonar ni de ganglios linfáticos hiliares. Recientemente se ha propuesto el término artritis granulomatosas pediátricas para unificar los desórdenes relacionados con mutaciones en CARD15 (el síndrome de Blau y la sarcoidosis de inicio temprano)11.
En este caso, las 2 primeras biopsias (separadas por un año) mostraron un infiltrado compuesto por histiocitos CD68+, CD1a– y S100–, sin clara formación de granulomas, compatible con una histiocitosis de células no LC, por lo que se diagnosticó inicialmente de forma errónea de HEG. Además, la presencia de células gigantes multinucleadas tipo Touton ayudó a confundir el diagnóstico. Clínicamente, nuestra paciente presentó pápulas con tendencia confluente y, a pesar de que con posterioridad se han publicado casos de HEG con lesiones confluentes formando placas, esto y su desaparición dejando cicatrices no concuerda con lo inicialmente descrito por Winkelmann y Muller en la HEG4.
Drummond et al.12 describen un caso similar en un hombre con sarcoidosis y presencia de histiocitos multinucleados, algunos con una corona de núcleos (tipo Touton), concluyendo que en algunos casos la prominencia de las células gigantes multinucleadas y el aspecto tipo Touton pueden sugerir diagnósticos alternativos, como xantogranulomas o xantoma diseminado, y demorar el diagnóstico, como en nuestro caso.
Los corticoides sistémicos siguen siendo de elección en el tratamiento de la sarcoidosis; estos están limitados por la aparición de efectos adversos. Por ello, y en casos refractarios, se han utilizado numerosos tratamientos alternativos, principalmente el metotrexato.
Hay evidencia de que el TNF-α está involucrado en la patogénesis de la sarcoidosis. No hay grandes estudios clínicos que comparen la eficacia entre los diferentes anti-TNF-α en niños con sarcoidosis. Existen solo casos aislados, series pequeñas y estudios clínicos limitados por el pequeño número donde los anti-TNF han demostrado ser eficaces en el tratamiento de sarcoidosis13,14.
Tanto infliximab15 como adalimumab15,16 han demostrado una mejoría rápida y marcada en lesiones cutáneas refractarias. Un trabajo controlado con infliximab en pacientes con sarcoidosis pulmonar demostró una modesta mejoría en el compromiso extrapulmonar17. En un estudio en niños con uveítis persistentemente activa y refractaria infliximab presentó mejores resultados que etanercept18.
En nuestra paciente, el uso prolongado de adalimumab (se mantuvo en total 36 meses) en combinación con metotrexato no presentó efectos adversos y permitió controlar el cuadro articular, pero estos fueron insuficientes para controlar totalmente el cuadro oftalmológico y las lesiones cutáneas, que mejoraron inicialmente, empeoraron posteriormente coincidiendo con reagudizaciones oculares. El cambio a infliximab permitió mantener la remisión de las lesiones cutáneas y articulares, e inducir una mejoría en el cuadro oftalmológico, lo que permitió disminuir la dosis de corticoides orales.
ConclusionesEn resumen, es importante siempre sospechar una sarcoidosis cuando nos enfrentamos a un niño con lesiones cutáneas asociadas a artritis y uveítis. A pesar de que la evidencia del uso de agentes anti-TNF-α en niños es todavía limitada, pueden ser una opción segura para el tratamiento de sarcoidosis refractaria. El uso de infliximab permitió inducir una mejoría en el compromiso oftalmológico, permitiendo disminuir la dosis de corticoides, mientras que adalimumab mostró una mejoría incompleta.
Conflicto de interesesLos autores declaran que no existe ningún conflicto de intereses.
