



Caso clínico
Motivo de consulta: niño de 8 años de edad remitido por episodios sincopales en los últimos 2 años, consistentes en pérdidas bruscas de conciencia sin aura ni signos vegetativos, de pocos minutos de duración y recuperación espontánea, siempre relacionados con ejercicio físico o emociones. Varios de los episodios se habían producido tras inmersión en piscina.
Antecedentes familiares: un tío paterno había fallecido a la edad de 30 años por muerte súbita de causa no aclarada. El padre había sufrido episodios similares a los del paciente durante la infancia, que desaparecieron posteriormente.
Antecedentes personales: sin interés.
Examen físico: sin alteraciones.
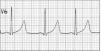
Exámenes complementarios: hemograma, glucemia e ionograma normales. Examen neurológico, electroencefalograma y tomografía computarizada craneal normales. En la figura 1 se muestra el trazado de la derivación V6 obtenido en el electrocardiograma (ECG).
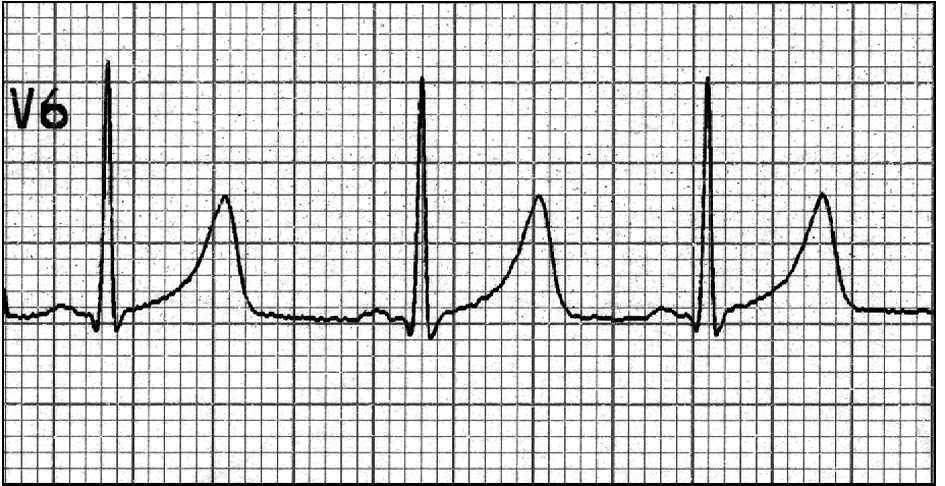
Figura 1. Trazado de la derivación V6.
Preguntas
1. ¿Cuál es el diagnóstico de presunción?
2. ¿Qué exámenes adicionales deben realizarse para confirmarlo?
Síndrome de intervalo QT largo congénito
El intervalo QT del trazado electrocardiográfico (medido desde el inicio de la onda Q hasta el final de la onda T) es de 0,42 s. Como el QT varía según la frecuencia cardíaca, los valores medidos deben normalizarse para ésta, para lo que se suele utilizar la fórmula de Bazett: QT corregido = QT medido/raíz cuadrada del intervalo RR. El QT corregido (QTc) se considera prolongado si excede los 0,44 s; en este caso era de 0,42/0,88 = 0,48 s.
Se realizaron: a) ECG-Holter de 24 h, en el que se detectaron valores de QTc anómalos en el 99 % del registro, que alcanzaron los 0,59 s, evidenciándose un aumento del QTc al aumentar la frecuencia cardíaca, y b) ergometría, en la que se constató un aumento progresivo del QTc con el esfuerzo, con un máximo de 0,57 s al final de la prueba; no se registraron disritmias (fig. 2). La ausencia de acortamiento del QTc al aumentar la frecuencia cardíaca detectada en estas pruebas junto con la medida patológica del QTc en el ECG basal, la clínica y los antecedentes familiares son diagnósticos de síndrome de QT largo1.
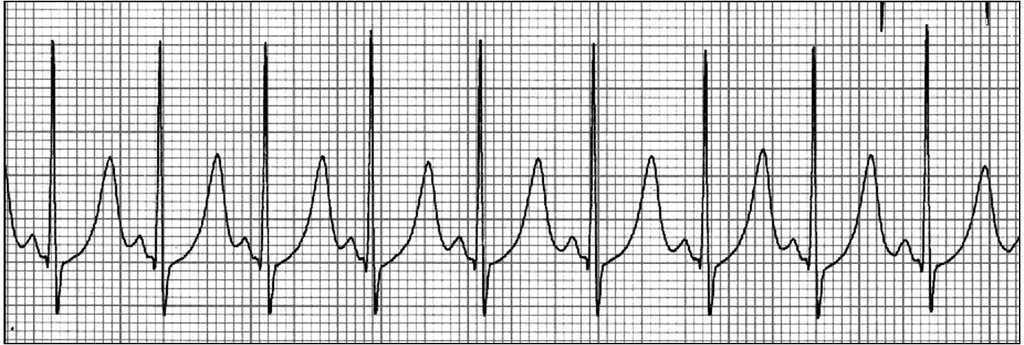
Figura 2. Ergometría.
El síndrome de QT largo congénito es una enfermedad hereditaria producida por anomalías en la función de los canales de sodio y potasio del corazón. El intervalo QT del ECG refleja la duración de las fases de despolarización y repolarización cardíacas, que en este proceso se encuentran enlentecidas. La prolongación del QT puede ocasionar arritmias ventriculares, típicamente episodios de taquicardia ventricular polimórfica con torsión de puntas (es decir, con cambios rápidos de polaridad que crean la impresión de que el trazado ECG se retuerce sobre sí mismo); estas taquiarritmias pueden ser autolimitadas o degenerar en fibrilación ventricular, produciendo la muerte súbita2-4.
El síndrome se caracteriza clínicamente por episodios sincopales que a menudo son precipitados por un incremento brusco de la actividad simpática (ejercicio físico, emociones); la inmersión es el desencadenante de los síncopes en al menos el 15 % de los casos. La tasa de muerte súbita es elevada después de la aparición del primer episodio sincopal, siendo la mortalidad a 10 años sin tratamiento de alrededor del 50 %4.
Existen dos grupos de intervalo QT largo congénito: el síndrome de Romano-Ward, de herencia autosómica dominante, para la cual se conocen actualmente cinco mutaciones de genes que codifican los canales iónicos miocárdicos, y el síndrome de Jerwell-Lange-Nielsen, asociado a sordera neurosensorial congénita; en este caso, el gen responsable del QT largo se hereda de forma autosómica dominante, mientras que la sordera lo hace de forma recesiva3-5.
Algunos casos de QT largo pueden ser secundarios al uso de fármacos que prolongan el intervalo QT (algunos antiarrítmicos, broncodilatadores, antibióticos macrólidos, antidepresivos tricíclicos y cisaprida, entre otros), a alteraciones electrolíticas (hipopotasemia, hipocalcemia, hipomagnesemia) o neurológicas (hemorragia subaracnoidea)4, que fueron descartados en este caso.
En la mayoría de variedades de intervalo QT largo congénito, la administración de bloqueantes b previene la aparición de síncopes y disminuye sustancialmente la tasa de muerte súbita. En este paciente se inició tratamiento con propranolol, encontrándose asintomático hasta el momento.
