






El síndrome de activación macrofágica (SAM) es una complicación grave de la artritis idiopática juvenil con forma de comienzo sistémico (AIJ-S).
ObjetivoDescribir las características clínicas y la evolución de los pacientes con SAM diagnosticados en las unidades de reumatología pediátrica en España.
Pacientes y métodoSe elaboró un protocolo de recogida de datos que se distribuyó a pediatras y reumatólogos que atienden niños con enfermedades reumáticas.
ResultadosSe recogieron datos de 31 pacientes (16 varones y 15 mujeres) que desarrollaron 37 episodios de SAM. Un total de 27 tuvieron un solo episodio; en 3 casos hubo dos episodios y 1 tuvo cuatro episodios, con intervalos entre ellos de 1 a 33 meses. La media de edad en el primer episodio fue 5,9 años (rango 1-23). El SAM fue la primera manifestación de AIJ-S en 9 casos. El síntoma más frecuente fue la fiebre (97 %), seguido de exantema (49 %), alteraciones neurológicas (41 %) y digestivas (15 %). Los datos analíticos más frecuentes fueron trombocitopenia (78 %) y aumento de las enzimas hepáticas (70 %). En 16 de 30 médulas óseas examinadas se objetivó hemofagocitosis. Todos los episodios menos uno fueron tratados con corticoides. Ciclosporina A fue utilizada en 15 episodios y etopósido, en seis. En un paciente se realizó un trasplante hepático previo al diagnóstico de SAM. El nivel de mortalidad fue del 6,5 % (2/31).
ConclusionesEl SAM es una complicación grave, potencialmente fatal, de la AIJ-S. Las alteraciones características de la enfermedad de base dificultan el diagnóstico. La disminución en el recuento de plaquetas y la elevación de las enzimas hepáticas fueron los hallazgos más frecuentes y precoces. Un alto grado de sospecha, un diagnóstico y un tratamiento precoces son esenciales para la resolución del cuadro.
Macrophage activation syndrome (MAS) is a severe complication of systemic juvenile idiopathic arthritis (sJIA).
ObjectiveTo describe the clinical characteristics and outcome of patients diagnosed with MAS in Spanish pediatric rheumatology units.
Patients and methodA protocol for data collection was designed and distributed to pediatricians and rheumatologists attending children with rheumatic diseases.
ResultsInformation was available from 31 patients (16 boys and 15 girls) who had 37 MAS episodes. Twenty-seven children had only one episode, three had two episodes and one had four episodes. The interval between episodes ranged from 1 to 33 months. The median age was 5.9 years (range 1-23). MAS was the initial manifestation of sJIA in nine patients. The most frequent symptom was fever (97 %), followed by skin rash (49 %), central nervous system dysfunction (41 %), and gastrointestinal abnormalities (15 %). Abnor-mal laboratory findings included thrombopenia (78 %) and elevated levels of hepatic enzymes (70 %). Hemophagocytosis was confirmed in 16 of 30 bone marrow samples evaluated, 15 with cyclosporine A and six with etoposide. All episodes but one were treated with steroids. One patient received a liver transplant before diagnosis. The mortality rate was 6.5 % (2/31).
ConclusionMAS is a severe, potentially lethal, complication of sJIA. The clinical and laboratory abnormalities characteristic of sJIA complicate its diagnosis. The earliest and most frequent findings were decreased in platelet count and elevation of hepatic enzymes. A high degree of suspicion as well as early diagnosis and prompt treatment are essential in this disease.
El síndrome de activación macrofágica (SAM) es una complicación grave de las enfermedades reumáticas infantiles y, en especial, de la artritis idiopática juvenil (AIJ) en su forma sistémica (AIJ-S)1–8. Se incluye en la linfohistiocitosis hemofagocítica (LHH) como se muestra en la tabla 19. La etiología es desconocida, aunque fármacos y virus se describen como posibles desencadenantes. En la patogenia está implicada una proliferación incontrolada de linfocitos T y de macrófagos que da lugar a una liberación excesiva de citocinas inflamatorias, responsables del daño tisular y de las alteraciones analíticas10. Se caracteriza clínicamente por fiebre, exantema, hepatoesplenomegalia y alteración del sistema nervioso central (SNC). En los análisis se encuentra pancitopenia, aumento de enzimas hepáticas y coagulopatía. Otros hallazgos habituales son la normalización y la disminución paradójica de la velocidad de sedimentación globular (VSG), el aumento de ferritina, triglicéridos y lactato deshidrogenasa (LDH). El dato anatomopatológico característico es la presencia de macrófagos bien diferenciados fagocitando células hematopoyéticas. El diagnóstico no es fácil debido a que algunos síntomas son comunes con la AIJ-S y las alteraciones hematológicas pueden estar ocultas por alteraciones previas debidas al proceso inflamatorio de base. El objetivo de este trabajo es describir los casos de SAM en nuestro país y analizar sus dificultades diagnósticas.
Clasificación de la linfohistiocitosis hemofagocítica9
| LHH genética o primariaLHH familiarDefectos genéticos conocidos: perforina, otrosDefectos genéticos desconocidosDéficit inmunológicosSíndrome de Chédiak-HigashiSíndrome de Griscelli Síndrome linfoproliferativo ligado al cromosoma XLHH adquirida o secundariaInfección (agentes infecciosos, toxinas): síndrome hemofagocítico asociado a infecciónProductos endógenos (daño tisular, productos metabólicos)Enfermedades reumáticas: síndrome de activación macrofágicaEnfermedades malignas |
LHH: linfohistiocitosis hemofagocítica.
Se elaboró un protocolo de recogida de datos que se distribuyó a pediatras y reumatólogos que atienden a niños con enfermedades reumáticas. Se incluyeron los pacientes con AIJ-S según los criterios establecidos por la International League of Associations for Rheumatology11 y aquéllos con AIJ-S “probable”, que se definió como aquélla en que los pacientes tuvieron fiebre en picos, persistente al menos durante 2 semanas, con estudios etiológicos negativos (infecciones, tumores, etc.) y que no habían presentado artritis o ésta había tenido una duración inferior a 6 semanas. Para el presente estudio, la enfermedad se consideró “activa” cuando había signos clínicos o analíticos de actividad inflamatoria durante el mes anterior al comienzo del SAM. Se definió “inactiva con tratamiento” si el paciente recibía tratamiento y estaba sin signos de actividad desde al menos un mes antes, e “inactiva sin tratamiento” si no había signos de actividad ni había requerido tratamiento el mes anterior. Se consideró el exantema signo clínico de SAM si era diferente al típico de la AIJ-S. Se definió “recidiva” cuando un nuevo episodio apareció al menos un mes después de la normalización clínica y analítica de un SAM previo. Dos casos se han descrito con anterioridad12,13. Los resultados se analizaron con el programa SPSS versión 11.0 mediante el test de Wilcoxon para datos pareados.
resultadosCaracterísticas generalesSe recogieron los datos de 31 pacientes, 16 varones y 15 mujeres. En total se estudiaron 37 episodios de SAM. Un total de 27 niños habían tenido un único episodio, 3 casos tuvieron dos episodios y otro niño tuvo cuatro. La media de edad fue de 5,9 años (rango 1–23). El diagnóstico de la enfermedad de base fue AIJ-S en 25 pacientes y AIJ-S “probable” en seis, sin que se apreciaran diferencias en las características del SAM entre ambos grupos. La AIJ-S estaba activa en 22 episodios, inactiva con tratamiento en cuatro e inactiva sin tratamiento en dos. En los restantes nueve episodios el SAM fue la primera manifestación de la AIJ-S. En la mayoría de los episodios (24/37) los pacientes tomaban una o varias medicaciones en el momento del desarrollo del SAM: 21 antiinflamatorios no esteroideos, 14 corticoides, 4 metotrexato, 3 etanercept y 3 anakinra. Tres habían recibido gammaglobulina intravenosa en el mes anterior y otro, sales de oro intramuscular12. Un total de 8 niños habían tomado antibióticos; 2 aciclovir, y otros 2, tuberculostáticos por sospecha de tuberculosis, que no se confirmó. Asimismo, 8 niños habían tomado al menos uno de los siguientes fármacos: tacrólimus, sulfadiazina, itraconazol o alendronato sódico. Un paciente con AIJ-S refractaria estaba recibiendo fludarabina y globulina antitimocítica como tratamiento acondicionador para trasplante autólogo de médula ósea (TMO) y a los 30 días del mismo presentó un nuevo episodio de SAM13. Se recogió el antecedente de infección del tracto respiratorio en siete episodios, gastroenteritis aguda en tres, mononucleosis infecciosa en dos, varicela en uno y herida infectada en otro. Un paciente tenía un síndrome nefrótico secundario a una amiloidosis por AIJ-S.
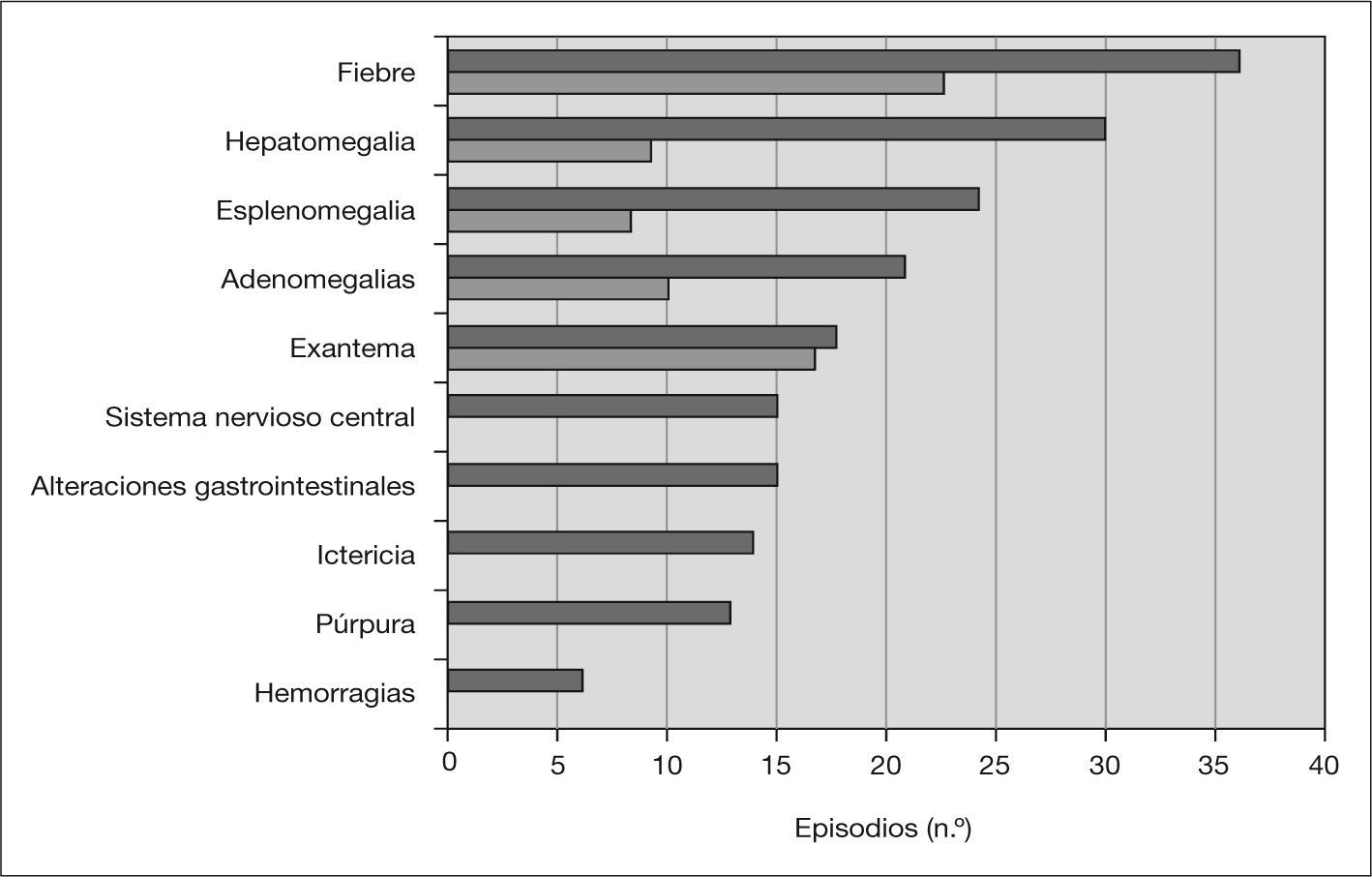
Manifestaciones clínicasLas manifestaciones clínicas del SAM y de la AIJ-S en el mes anterior se muestran en la figura 1. El síntoma más frecuente fue la fiebre (97%), seguido de exantema (49%), alteraciones neurológicas (41%) y digestivas (15%). El exantema fue descrito como fijo y generalizado en la mitad de los casos y con componente purpúrico el 42 % de las veces. Las manifestaciones neurológicas fueron alteración de la conciencia en 11 episodios y convulsiones en cuatro. Dos pacientes tuvieron hemorragia gingival; otro, melenas; otro, rectorragia, y dos, epistaxis. Un niño presentó hematuria macroscópica. Las alteraciones digestivas fueron vómitos en doce ocasiones y diarrea en siete. Se objetivó hepatomegalia en el 81 % de los episodios y esplenomegalia, en el 65 %. Se detectó derrame pleural en 4 casos, pericarditis en 2, ascitis en otros 2 y pancreatitis con desarrollo posterior de seudoquiste en uno.
Los datos analíticos previos al SAM, al inicio del mismo y durante la evolución se muestran en la tabla 2. Los leucocitos fueron ≤ 4 × 109/l en 8 episodios y los neutrófilos fueron ≤ 1 × 109/l en 3. Las plaquetas estuvieron por debajo de 150 × 109/l en 29 episodios y la hemoglobina, ≤ 9g/l en 19. Dado que muchos pacientes partían de cifras muy altas de leucocitos y de plaquetas debido a la AIJ-S, se calculó el porcentaje de disminución de dichos parámetros, que fue del 42 % para los leucocitos, del 39 % para los neutrófilos y del 71 % para las plaquetas. En 10 episodios se encontró un aumento de los productos de degradación del fibrinógeno. Un niño a quien se le realizó punción lumbar presentó pleocitosis. La VSG disminuyó un 58% en la mitad de los episodios. Las transaminasas eran ≥ 100 Ul/ml en 29 episodios y el tiempo de protrombina estuvo por debajo del 63 % en la mitad de ellos. La ferritina, previamente elevada en muchos pacientes, subió espectacularmente en algunos casos. La actividad citotóxica de las células natural killer (NK) y la expresión de perforina fue normal en un paciente en el que se estudió y el nivel del receptor soluble de la cadena alfa de la IL-2 (sCD25) en ese caso fue de 3.556pg/ml (normal: 0–1.900).
Datos de laboratorio en 37 episodios de síndrome de activación macrofágica antes del comienzo del mismo, al inicio y durante la evolución
| Pre-SAM | Inicio del SAM | p** | Evolución | p*** | |
| Media (rango intercuartílico) | Media (rango intercuartílico) | Media (rango intercuartílico) | |||
| Leucocitos (× 109/l)* | 14,2 (8,7-23,9) | 9,5 (5,2-16,8) | 0,004 | 6 (3,9-8,5) | < 0,001 |
| Neutrófilos (× 109/l)* | 10 (4,9-20,1) | 6 (2,5-10) | 0,010 | 3,2 (1,4-5,7) | 0,001 |
| Plaquetas (× 109/l)* | 376 (283-515) | 102 (69-158) | < 0,001 | 84 (45-153) | < 0,001 |
| Hemoglobina (g/dl)* | 11,2 (10-12) | 9 (8,2-10,3) | < 0,001 | 8,8 (7,5-10) | < 0,001 |
| Ferritina Cμg/l) | 846 (212-2.867) | 4.102 (999-20.000) | 0,017 | 4.680 (1.374-34.304) | 0,068 |
| VSG (mm/h) | 71 (33-95) | 23 (15-43) | 0,003 | 24 (15-41) | 0,008 |
| PCR (mg/l) | 78 (7,5-135) | 78 (36-109) | 0,435 | 17 (8-71) | 0,203 |
| Proteínas (g/l) | 6,7 (6,5-7,5) | 5,8 (5-6,2) | < 0,001 | 5,5 (4,7-6,7) | 0,037 |
| Albúmina (g/l) | 3,4 (2,9-3,7) | 2,8 (2,5-3,3) | 0,075 | 2,5 (2,1-2,9) | 0,500 |
| Triglicéridos (mg/dl) | 96 (56-146) | 284 (136-400) | 0,043 | 321 (189-434) | 0,080 |
| LDH (UI/l) | 471 (384-717) | 1.292 (786-3.904) | < 0,001 | 1.227 (646-6.556) | 0,012 |
| Bilirrubina (mg/dl) | 0,4 (0,3-0,6) | 1,5 (0,5-3,1) | 0,001 | 3,1 (1,1-6,7) | 0,017 |
| GPT (UI/l) | 15 (9-28) | 210 (79-476) | < 0,001 | 218 (64-409) | 0,002 |
| Protrombina (%) | 80 (74-89) | 63 (46-78) | 0,049 | 64 (34-91) | 0,345 |
| Fibrinógeno (g/l) | 5,3 (2,2-6,9) | 1,8 (1-3,2) | 0,026 | 2,3 (0,9-3,6) | 0,183 |
GPT: transaminasa glutámica pirúvica; LDH: lactato deshidrogenasa; PCR: proteína C reactiva; SAM: síndrome de activación macrofágica; VSG: velocidad de sedimentación globular.
En 14 de 30 aspirados y en 4 de 9 biopsias de médula ósea se encontró hemofagocitosis. Los resultados entre el aspirado y la biopsia no siempre concordaron. Se realizaron 5 biopsias hepáticas y 4 ganglionares; sólo en una de cada se objetivó hemofagocitosis y en ambos casos ya había sido encontrada en la muestra de médula ósea. En total, la hemofagocitosis se observó en 16 episodios. El número de criterios diagnósticos varió entre 1 y 6 de los establecidos por la Sociedad Histiocítica (tabla 3)9 y entre 2 y 8 considerando los propuestos por Ravelli et al14 (tabla 4).
Criterios diagnósticos de linfohistiocitosis hemofagocítica9
| Enfermedad familiar o defecto genético conocidoCriterios clínicos y de laboratorio (5 de 8 criterios)FiebreEsplenomegaliaCitopenia en dos o más líneas celulares:Hemoglobina < 9g/dlPlaquetas < 100 × 109/lNeutrófilos < 1 × 109/lHipertrigliceridemia ≥ 3mmol/l en ayunas o hipofibrinogenemia < 1,5g/lFerritina ≥ 500μg/lsCD25 ≥ 2.400 UI/mlDisminución o ausencia de la actividad de las células NKHemofagocitosis en médula ósea, LCR o ganglios |
Los siguientes datos apoyan el diagnóstico: síntomas cerebrales con moderada pleocitosis y/o proteínas elevadas, aumento de las transaminasas, bilirrubina y lactato deshidrogenasa.
LCR: líquido cefalorraquídeo; NK: natural kíller; sCD25: receptor soluble de la cadena alfa de la interleucina-2.
Criterios diagnósticos del síndrome de activación macrofágica en la artritis idiopática juvenil sistémica14
Criterios de laboratorioDisminución del recuento de plaquetas (≤ 262 × 109/l)Aumento de GPT (> 59 Ul/l)Disminución de los leucocitos (≤ 4,00 × 109/l)HipofibrinogenemiaCriterios clínicosDisfunción del SNC (irritabilidad, desorientación, letargia, cefalea, convulsiones, coma)Hemorragias (púrpura, sangrado gingival, etc.)Hepatomegalia (≥ 3cm por debajo del reborde costal)Criterio histopatológicoEvidencia de hemofagocitosis macrofágica en el aspirado de médula óseaReglas diagnósticasEl diagnóstico de SAM requiere la presencia de dos o más criterios de laboratorio o dos o más criterios clínicos o de laboratorioUn aspirado de médula ósea para demostrar hemofagocitosis es necesario solamente en casos dudososRecomendacionesLos criterios referidos sólo son válidos para pacientes con AIJ-S.Los valores entre paréntesis sólo son dados como ejemploComentarios
|
AIJ-S: artritis idiopática juvenil sistémica; GPT: transaminasa glutámica pirúvica;
LDH: lactato deshidrogenasa; SAM: síndrome de activación macrofágica;
SNC: sistema nervioso central; VSG: velocidad de sedimentación globular.
Todos los episodios salvo uno fueron tratados con corticoides, 21 en forma de bolos intravenosos (i.v.) de metilprednisolona (MP) a 10–30mg/kg/día. Se utilizó ciclosporina A (CyA) en 15 episodios, inicialmente i.v. en 11 casos, en dosis de 2–8mg/kg/día. En seis episodios (5 pacientes) se utilizó etopósido a 150/mg/m2 y el número de dosis varió entre 3 y 15. La respuesta se consideró buena en todos. Otros tratamientos fueron hemofiltración en tres episodios, plasmaféresis en dos, tacrólimus en dos, gammaglobulina i.v. en uno y anakinra en otro. En 15 ocasiones fue necesaria la estancia en cuidados intensivos.
Un niño que había tenido convulsiones durante el SAM volvió a presentarlas un mes después de controlado el proceso, sin otros síntomas acompañantes. Un paciente presentó un SAM estando en tratamiento acondicionador para un TMO. Tras su recuperación, y al mes del TMO, presentó una recidiva. En otro paciente se realizó un trasplante hepático antes de conocerse el diagnóstico. Nueve meses después presentó un nuevo episodio. En la actualidad está asintomático tras 2 años de seguimiento. En total, 4 pacientes tuvieron seis recidivas, en un tiempo que osciló entre 1 y 33 meses tras la recuperación del primer episodio (media de 10 meses). Dos pacientes murieron, uno de ellos a los 10 días, que además tenía una amiloidosis secundaria a la AIJ-S con insuficiencia renal; el otro falleció a los 2 días del ingreso por un fallo multiorgánico. El SAM se resolvió en 35 episodios en un período que osciló entre los 3 y los 180 días (media de 20 días).
discusiónEl SAM es una complicación grave de la AIJ-S que se encuadra dentro de la LHH secundaria. La LHH es un término general que incluye un espectro de enfermedades producidas por proliferación y activación de linfocitos T y macrófagos que conduce a una respuesta inflamatoria excesiva con hipersecreción de citocinas como el interferón gamma (IFN-γ), el factor de necrosis tumoral alfa (TNF-α), interleucina 1 (IL-1), IL-6, IL-10, IL-12, IL-18 y el factor estimulante de las colonias de macrófagos, responsables del daño tisular y de las alteraciones analíticas10,,15–17. Se clasifica en genética o primaria y adquirida o secundaria (tabla 1)9,15,18. La LHH primaria suele manifestarse en el primer año de vida. La LHH secundaria se presenta en infecciones, tumores y enfermedades reumáticas, y en este caso se le denomina SAM. Las infecciones más frecuentes que se asocian a LHH son las producidas por el virus de Epstein-Barr, citomegalovirus y herpes virus, de ahí la denominación de virus-associate haemophagocytic syndrome o VAHS, pero también se ha observado en infecciones por bacterias, protozoos y hongos, por lo que el término más correcto sería el de síndrome hemofagocítico asociado a infección (SHAI). En la LHH secundaria a enfermedades malignas y reumáticas dichas infecciones desempeñan potencialmente un papel desencadenante.
Los mecanismos patogénicos involucrados en el SAM no se conocen. En la LHH familiar la proliferación incontrolada de células T y macrófagos está asociada con disminución de la actividad de las células NK y de los linfocitos T citotóxicos, secundario a mutaciones del gen de la perforina (PRF1)19,20. En pacientes con SAM asociado a AIJ-S se han encontrado alteraciones inmunológicas similares10. Bleesing et al21 encuentran valores elevados del receptor alfa soluble de la IL-2 y del CD163 soluble durante la fase aguda del SAM, que se normalizan al resolverse el cuadro. Wulffraat et al22 indicaron una disminución de la expresión de perforina en las células NK y en linfocitos T citotóxicos en niños con AIJ-S comparados con los que tienen otra forma de AIJ. Estos datos parecen sugerir que la disminución de la función de las NK asociada a valores anormales de expresión de perforina puede ser un dato que diferencie a pacientes con AIJ-S de los afectados por otras formas de AIJ y que explique la mayor incidencia del SAM en las primeras23.
El cuadro clínico del SAM comienza de forma aguda, con fiebre persistente, cambios del estado mental, linfadenopatía, hepatoesplenomegalia, facilidad para hematomas y sangrado de mucosas. Estos signos se asocian con una disminución de la cifra de plaquetas, leucocitos y hemoglobina. Las enzimas hepáticas están habitualmente aumentadas y a veces se observa hiperbilirrubinemia y disminución de la albúmina. La hepatopatía está inducida por la infiltración por macrófagos. La coagulopatía es la alteración más importante, y se observan alargamiento del tiempo de protrombina, del tiempo parcial de tromboplastina, hipofibrinogenemia y déficit de factores de la coagulación1–8,24–26. Es secundaria a la afectación hepática, aunque pueden contribuir a su desarrollo un grado de coagulopatía de consumo27 y el incremento de la actividad del TNF-α encontrado en la AIJ-S28. Una caída en la VSG es otro dato característico. Otras alteraciones son las concentraciones elevadas de triglicéridos, LDH y ferritina. El SAM puede ser la primera manifestación de la AIJ-S, lo que dificulta el diagnóstico29. Anteriormente se ha hecho mención a la posibilidad de que se presenten recidivas5,7,30.
Se desconoce cuál es la incidencia del SAM. Los criterios diagnósticos utilizados para la LHH (v. tabla 3)9 son difíciles de aplicar por varias razones la fiebre es un síntoma y un criterio tanto de AIJ-S como de LHH, los niños con AIJ-S suelen presentar cifras muy elevadas de leucocitos, plaquetas y fibrinógeno, por lo que cuando desarrollan un SAM, la leucopenia, la trombocitopenia y la hipofibrinogenemia pueden ser relativas. La hemofagocitosis no siempre se encuentra y su búsqueda, en ocasiones, puede demorar el diagnóstico y el tratamiento. El estudio de la actividad de las células NK y del valor del sCD25 es laborioso y requiere un laboratorio altamente especializado. Por todo ello, Ravelli et al14, tras un estudio en el que se comparaba pacientes con brote de actividad de AIJ-S con otros con SAM, proponen unos criterios preliminares (v. tabla 4), insistiendo en la necesidad de validarlos prospectivamente.
El objetivo del tratamiento es frenar el proceso hiperinflamatorio. Para una normalización rápida de las alteraciones analíticas se recomiendan bolos i.v. de MP (30mg/kg/día durante 3 días consecutivos) seguidos por 2–3mg/kg/día en 3 o 4 dosis. Tras la normalización de las alteraciones hematológicas y la resolución de la coagulopatía, la disminución de los corticoides debe hacerse lentamente para evitar reactivaciones4,10,18. Con cierta frecuencia el proceso es resistente a los corticoides, y se ha descrito el caso de muertes incluso en pacientes tratados con dosis masivas de corticoides. En estos casos se debe asociar CyA oral o parenteral a dosis de 2–8mg/kg/día31,32. Hasta ahora los casos tratados con etopósido, un derivado del alcaloide podofilotoxina, son anecdóticos; sin embargo, la mayor experiencia con este compuesto en el tratamiento de la LHH familiar33 hace razonable su uso en pacientes con SAM grave que requieran dosis altas de corticoides, aunque la pauta de administración se adapte a la respuesta del paciente. También se ha usado inmunoglobulina i.v.34. Los receptores solubles del TNF y de la IL-1 (etanercept y anakinra, respectivamente) se han utilizado con una buena respuesta en algunas ocasiones, pero paradójicamente también se han descrito como desencadenantes35–39. Otras medidas importantes son la investigación y el tratamiento de posibles infecciones asociadas, así como la retirada de fármacos que puedan favorecer el proceso. Un adecuado tratamiento de soporte es imprescindible en situaciones graves.
En este estudio se revisan las características de 37 episodios de SAM ocurridos en 31 niños. El proceso se presentó con igual frecuencia en ambos sexos y el rango de edad fue muy amplio. En el 38% de las ocasiones los pacientes habían tenido una infección y en el 65 % estaban tomando alguna medicación. El valor de estos factores como desencadenantes o la posibilidad de que sea una coincidencia son difíciles de determinar. El SAM fue la primera manifestación de la AIJ-S en el 29 % de los pacientes en este estudio, mientras que en la serie de Stéphan et al5 esto sucedió en el 12,5% y en la de Kounami et al7, en el 40%. La mayoría de los pacientes tuvieron fiebre, al igual que en el resto de los casos publicados. Tanto los médicos como los padres refirieron que el exantema era diferente al típico de la AIJ-S. Este dato está poco descrito, probablemente debido a la dificultad para diferenciarlo de la erupción cutánea característica de la AIJ-S. Las manifestaciones del SNC se encontraron en el 41 % de los episodios, similar a la referida por otros autores5,7,14, y la alteración del estado mental –en concreto, la somnolencia– fue uno de los síntomas iniciales más frecuentes. En la bibliografía hay pocas referencias a las manifestaciones digestivas no hemorrágicas, pero en nuestro estudio se encontraron en el 15 % de los episodios. La hepatomegalia, la esplenomegalia y las adenomegalias fueron los signos clínicos más observados, pero, como muestra la figura 1, estaban presentes en un porcentaje de pacientes antes del inicio del SAM, lo que disminuye su poder discriminativo. Los leucocitos, neutrófilos y plaquetas, aunque con frecuencia dentro de parámetros normales, presentaron una disminución importante en relación con los valores previos al SAM, dando lugar a una “citopenia relativa”. La VSG, elevada en los controles previos, mostró a menudo una “mejoría paradójica”. Las enzimas hepáticas y la LDH estuvieron elevadas en la mayor parte de los episodios al igual que en el resto de las series. Sólo en un caso se estudió la actividad de las células NK y la expresión de perforina, con resultados normales. El valor del sCD25 en ese mismo paciente estuvo por encima del rango normal. El 13 % de los pacientes presentaron recidivas, algo menos que las comunicadas por otros autores5,7.
En todos los episodios estaban presentes al menos dos criterios de SAM asociado a AIJ-S propuestos por Ravelli et al14, pero sólo 11 reunían cinco criterios de los exigidos por la Sociedad Histiocítica para el diagnóstico de LHH9.
Todos los episodios, menos uno, fueron tratados con corticoides. Se añadió CyA cuando el paciente no respondió adecuadamente o desde el comienzo si la gravedad del cuadro lo aconsejaba. En seis episodios se utilizó etopósido en dosis y frecuencia variables, y se consideró que la respuesta fue buena en todos. Dos pacientes fallecieron; uno tenía una situación clínica mala, con un síndrome nefrótico secundario a amiloidosis, y el otro falleció a las 48h de su ingreso a pesar del tratamiento intensivo. El paciente que recibió un trasplante hepático cursó como insuficiencia hepática grave sin otros signos de linfohistiocitosis. La evolución final fue buena en el 90 % de los pacientes con recuperación total de los parámetros analíticos.
En resumen, el presente estudio es una revisión de los SAM, diagnosticados por pediatras y reumatólogos habituados al tratamiento de pacientes con enfermedades reumáticas infantiles, con el objetivo de documentar la experiencia conseguida. Como conclusión, consideramos que, en un paciente con AIJ-S, una disminución del recuento de plaquetas, una elevación de enzimas hepáticas y una caída de la VSG deben hacer sospechar un SAM inminente, y el tratamiento rápido e intensivo puede prevenir el desarrollo del proceso a gran escala.
Grupo de Estudio del Síndrome de Activación Macrofágica y Artritis Idiopática JuvenilInmaculada Calvo Penadés (Hospital La Fe, Valencia); Purificación Moreno Pascual (Hospital Carlos Haya, Málaga); Consuelo Modesto Caballero (Hospital Vall d'Hebron, Barcelona); M.ª Jesús Rúa Elorduy (Hospital de Cruces, Bilbao); Jordi Antón López (Hospital de Sant Joan de Déu, Barcelona); Pilar Solís Sánchez (Hospital Clínico-Universitario, Valladolid); Silvia Rodríguez Rubio, M.ª Luz Gámir Gámir (Hospital Ramón y Cajal, Madrid); Soledad Camacho Lovillo (Hospital Virgen del Rocío, Sevilla); Rosa Bou Torrent (Hospital Parc Taulí, Barcelona); Mercedes Ibáñez Rubio, M.ª Dolores López Saldaña (Hospital del Niño Jesús, Madrid); M.ª Antonia Carballo Silva (Hospital Xeral Cies, Pontevedra); Sagrario Bustabad Reyes, Juan-José Bethencourt Baute (Hospital Universitario de Canarias).
