


Sr. Editor:
El síndrome de Aicardi-Goutieres, descrito por primera vez en 19841, es una encefalopatía progresiva de inicio precoz con herencia autosómica recesiva2. Hasta el momento se han publicado unos 30 casos en Europa. La clínica se inicia a las pocas semanas de vida con irritabilidad, vómitos y rechazo del alimento. Posteriormente aparece retraso psicomotor grave. La tomografía computarizada (TC) craneal muestra calcificaciones y en el líquido cefalorraquídeo (LCR) se debe determinar el interferón α (IFN-α), que estará aumentado3.
Ante calcificaciones cerebrales prenatales o precoces hay que descartar infecciones congénitas, pero también se debe pensar en síndromes neurodegenerativos menos frecuentes como el síndrome de Aicardi-Goutieres, por lo que hemos considerado de interés la presentación de este caso clínico.
Recién nacido de sexo femenino con calcificaciones intracraneales detectadas prenatalmente.
Antecedentes familiares sin interés y sin consanguinidad entre los padres.
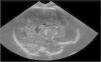
Antecedentes obstétricos: primera gestación controlada y sin patología materna. Serologías en el primer trimestre negativas-inmunes. Ecografías fetales inicialmente normales salvo crecimiento fetal en percentil 5 hasta la semana 37, en la que aparecen calcificaciones múltiples en los tálamos y la zona periventricular (fig. 1) y leve hepatoesplenomegalia. Se realiza resonancia magnética (RM) fetal que muestra aumento de densidad en núcleos de la base sin otras anomalías estructurales. Se repiten serologías maternas (toxoplasmosis, rubéola, citomegalovirus, herpes [TORCH] y parvovirus B19) que resultan de nuevo negativas-inmunes, sin signos de infección reciente. El cultivo vaginal y el rectal son negativos para Streptococcus agalactiae. El parto fue de inicio espontáneo, finalizó mediante cesárea de recurso en la semana 40 de gestación por sospecha de pérdida de bienestar fetal. Amniorrexis artificial con aguas meconiales. Nace niña con Apgar de 7/9/9.
Exploración física: peso 2.570g (p5-10), talla 46cm (p5), perímetro cefálico 32cm (p10). La exploración está dentro de la normalidad.
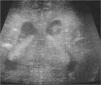
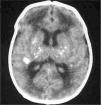
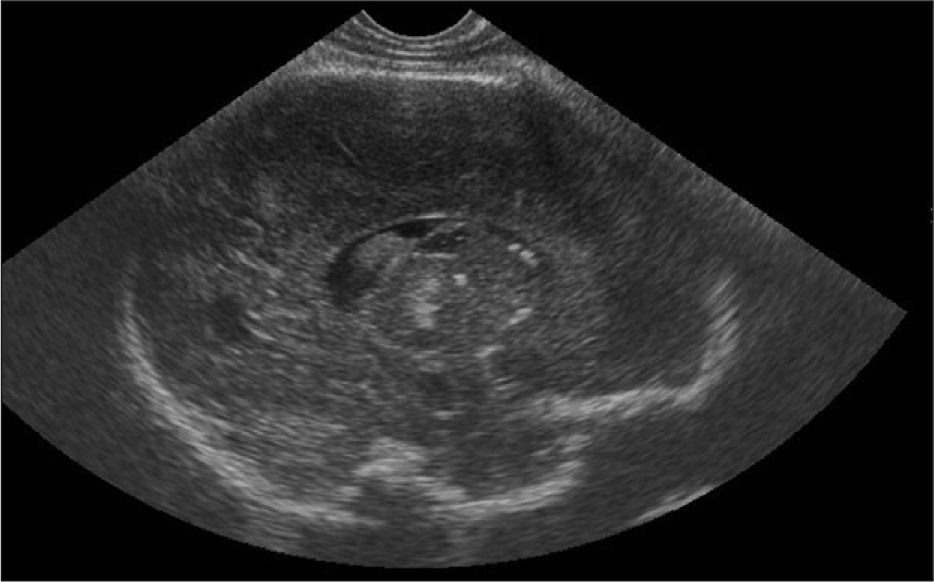
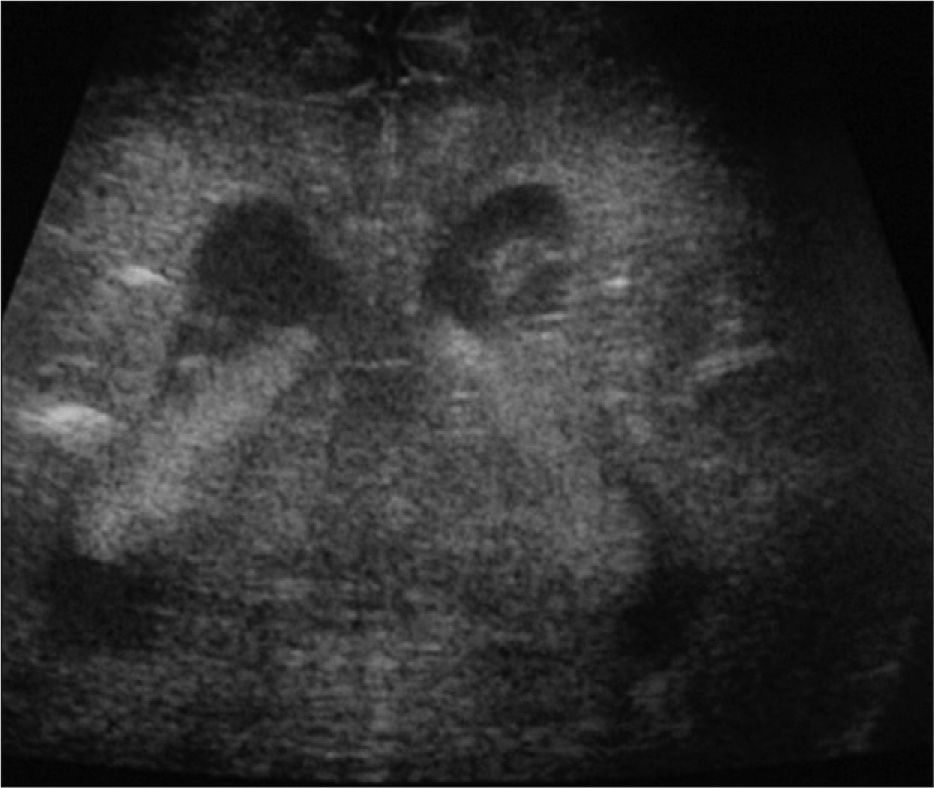
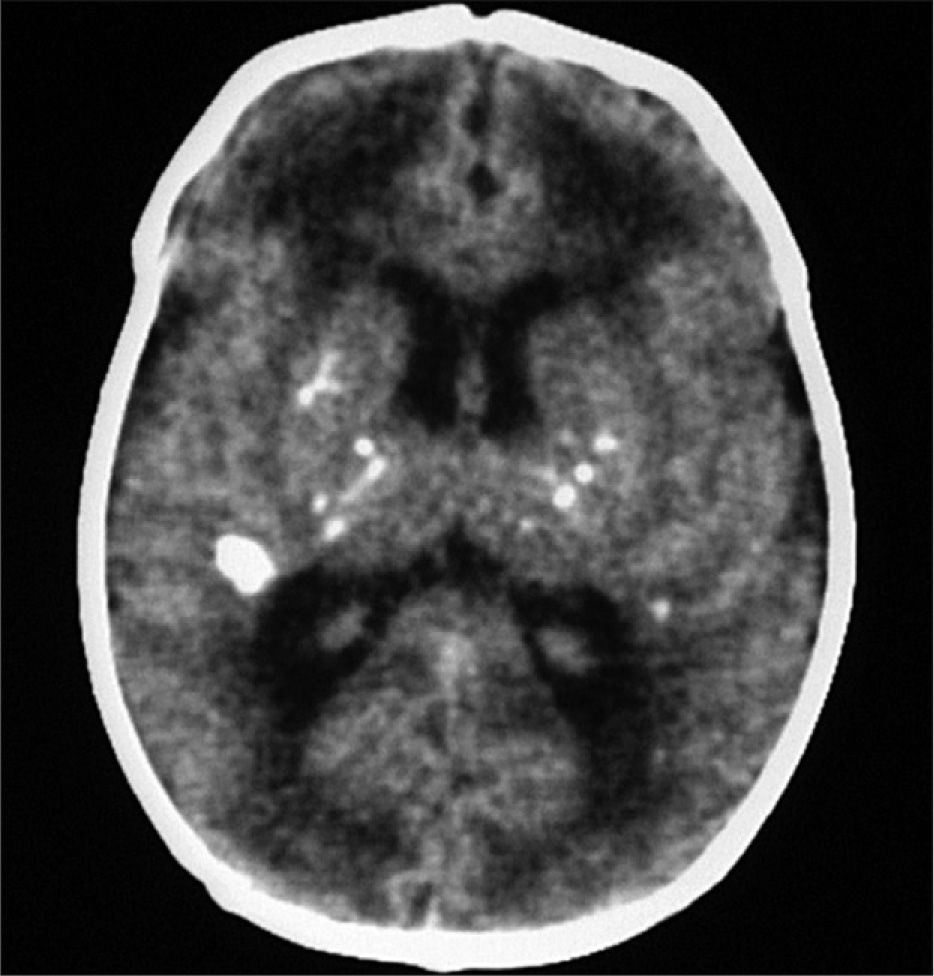
Exámenes complementarios: analítica sangre: 17.500 leucocitos (70 % granulocitos, 10 % linfocitos); hemoglobina, 13g/l; plaquetas, 153.000/μl; proteína C reactiva, 13mg/l; transaminasa glutámico oxalacética (GOT), transaminasa glutámico pirúvica (GPT), calcio, fósforo, amonio, láctico, hormona paratiroidea (PTH) y perfil tiroideo sin alteraciones. Los aminoácidos y los ácidos orgánicos en orina son normales. El citomegalovirus (CMV) en orina es negativo. Serologías toxoplasma y rubéola IgG+/IgM–. Lúes, herpes, parvovirus B19 y CMV IgG–/IgM–. Ecografía cerebral (fig. 2): calcificaciones múltiples en ambos hemisferios con afectación talámica. TC cerebral con contraste (fig. 3): calcificaciones múltiples en ambos hemisferios afectando sustancia blanca subcortical y sustancia gris profunda. Punción lumbar: linfocitosis y aumento de IFN-α y de pterinas, por lo que se estableció el diagnóstico de síndrome de Aicardi-Goutiéres. Se envió muestra para estudio genético.
Se le dio el alta a los 12 días de vida con exploración física y neurológica normal.
A los 2 meses de vida inicia irritabilidad, rechazo del alimento y escaso incremento del perímetro cefálico. Posteriormente, su tono cefálico es deficiente, con nistagmo, espasticidad y movimientos distónicos de extremidades y deterioro neurológico progresivo. Fallece a los 7 meses.
Estos pacientes presentan encefalopatía grave y progresiva de inicio precoz con disfunción piramidal y extrapiramidal (hipotonía de tronco, espasticidad de miembros, movimientos distónicos, discinesias orofaciales, nistagmo con ausencia de contacto visual). Aunque existe variabilidad intrafamiliar de gravedad4, el pronóstico es malo.
En el LCR se objetiva linfocitosis, moderada hiperproteinorraquia y elevación de IFN-α. que estará más aumentado que a nivel sistémico3. El IFN-α interviene en los mecanismos de defensa antivirales y antitumorales5 y no se detecta en sangre de sujetos sanos. Se ha descrito elevado en infecciones del sistema nervioso central como encefalitis herpética o por virus de la inmunodeficiencia humana (VIH), en meningitis virales (enterovirus, parotiditis, virus varicela-zóster y rubéola congénita) y en una única entidad no infecciosa como el neurolupus. No atraviesa la barrera hematoencefálica, por lo que si está aumentado en el LCR, la producción es intratecal por infección o, como en nuestro caso, por mala regulación debido a la mutación de su gen. El aumento de IFN-α como mecanismo proinflamatorio da lugar a una vasculitis y ésta a las calcificaciones cerebrales1 que se objetivan en la TC craneal: calcificaciones en ganglios de la base que suelen ser bilaterales y simétricas, leucodistrofia y atrofia periventricular y periférico cerebral6.
Se ha descrito una variante del síndrome con aumento de pterinas7 y descenso de folatos en LCR. Un aumento de producción de pterinas a nivel intratecal sería la primera alteración bioquímica y el tratamiento con ácido folínico en ensayos clínicos muestra mejoría clínica en algunos de los pacientes.
El diagnóstico prenatal2 es difícil por la hetereogenicidad genética. En la mitad de las familias8 se ha identificado una mutación en el cromosoma 3p21 que codifica el gen TREX1, también denominado DNAasa III, cuyas mutaciones afectan a la degradación de moléculas de ADN parcialmente reparadas con la consiguiente acumulación de estas moléculas que condicionan una respuesta inmunológica inadecuada.
El diagnóstico diferencial debe establecerse con infecciones congénitas (TORCH, CMV, VIH), enfermedades neurodegenerativas y endocrinometabólicas que cursen con calcificaciones intracraneales.
No existe un tratamiento causal para el síndrome de Aicardi-Goutiéres, aunque estudios genéticos basados en la regulación de los genes del IFN podrían aportar tratamientos en un futuro y consejo genético9.
