

El síndrome de Angelman se caracteriza por retraso mental, epilepsia, déficit del lenguaje, dismorfia facial y un fenotipo conductual característico. Los criterios clínicos diagnósticos están definidos por consenso desde 1995. Está causado por el déficit o inactivación del gen UB3A. Se describen varios tipos de alteraciones genéticas. Existe un porcentaje de casos que, cumpliendo los criterios diagnósticos, los estudios genéticos son negativos.
Consideramos necesario analizar las características de nuestros pacientes y las posibles correlaciones fenotipo-genotipo.
Material y métodosSe incluyeron todos los casos tratados en la unidad de neurología que cumplieron los criterios diagnósticos, durante el período 1981-2007. Para el diagnóstico genético, se efectuó un análisis de metilación e hibridación fluorescente in situ.
ResultadosSe estudió a 13 pacientes, 9 con estudio genético positivo y 4 con genética negativa que cumplieron criterios clínicos. La edad media de diagnóstico fue de 37 meses. Once casos presentaron microcefalia adquirida. Un occipucio plano, malformaciones bucales y maxilares, hipopigmentación, apariencia feliz e hiperactividad fueron unas características prácticamente constantes. Tanto el lenguaje como la marcha fueron las áreas que presentaron un mayor déficit. Doce casos presentaron epilepsia. Tres de los casos con estudio genético normal presentan menos microcefalia y mejor desarrollo psicomotor, sobre todo en la marcha.
ConclusionesEs necesario conocer las características fenotípicas del síndrome para solicitar un estudio genético específico y para establecer el diagnóstico en los casos con genética negativa. En nuestros pacientes se constató el fenotipo característico que describió Angelman. Los casos de deleciones presentaron una mayor gravedad y una peor evolución.
Angelman syndrome is characterised by mental retardation, epilepsy, speech impairment, facial dysmorphism and a characteristic behavioural phenotype. Diagnostic clinical criteria have been defined by consensus since 1995. It is caused by deficiency or inactivation of the UB3A gene. There is a percentage of cases which satisfy these clinical features but have negative genetic testing.
We consider it necessary to analyse the patient characteristics and possible phenotypegenotype correlations.
Material and methodsAll cases which were treated between 1981 and 2007 in a neurology unit and fulfilled the clinical criteria were included. Genetic diagnosis was made by methylation testing and fluorescent in situ hybridization.
ResultsThirteen patients were studied, nine with positive genetic testing and four with negative testing who completed the clinical criteria. The average age at diagnosis was 37 months. Eleven cases showed acquired microcephaly. Flat occiput, mouth and maxillary malformations, hypopigmentation, a happy appearance and hyperactivity were practically constant characteristics. Speech and walking ability were the areas which showed most deficit. Twelve cases had epilepsy. Three of the cases with normal genetic testing showed less microcephaly and better psychomotor development, particularly in walking ability.
ConclusionsThe phenotypical characteristics of the syndrome should be known before requesting specific genetic testing and to make a diagnosis even in cases with negative genetic. The phenotype characteristics that describe Angelman syndrome were verified. Deletion cases had a worse outcome.
El síndrome de Angelman (SA) es una enfermedad de base genética, causada por anomalías que afectan a un único gen de expresión materna, el gen UB3A1. El tipo de alteraciones genéticas que inactivan la región 15q11-q13 son las siguientes: a) deleción de 4-6 megabases (70-75 %); b) microdeleciones (< 1 %) que afecten al centro imprinting (CI) o al área donde reside el gen UB3A; c) reorganizaciones cromosómicas (< 1 %) del tipo translocaciones o inversiones; d) disomía uniparental paterna (DUP) entre 2 y 5 %; e) defectos del CI (1-5 %), y f) mutaciones intragénicas relacionadas con la metilación del gen UB3A (10-15 %). Existe un 10-15 % de los casos que cumplen los criterios diagnósticos, pero los estudios genéticos no revelan ninguna alteración, incluyendo resultados negativos en la prueba del gen UB3A2.
Los niños con SA se caracterizan por hipopigmentación, dismorfia craneofacial (microcefalia, prognatismo, protusión lingual), marcha espasmódica, lenguaje prácticamente nulo, risa inmotivada, hipermotricidad, retraso mental grave y convulsiones. Las características físicas y conductuales están perfectamente reflejadas en los criterios diagnósticos, no así el fenotipo crítico. Los criterios diagnósticos fueron definidos por consenso en 19953; en marzo de 2006 se publicó su actualización2.
Existe una gradación de gravedad en el fenotipo según la causa genética, a pesar de la gran variabilidad dentro de cada grupo4. Las de mayor gravedad se asocian a deleciones, mutaciones de gen UB3A y SA con genética negativa. Los casos menos graves se asocian a DUP o defectos del CI5,6.
Las principales entidades con las que se plantea el diagnóstico diferencial incluyen el síndrome de Lenox-Gastaut, el síndrome de Rett, la parálisis cerebral atáxica de etiología indeterminada, deleciones terminales de la región 22q13, síndrome de Prader-Willi, síndrome de Mowat y algunas encefalopatías mitocondriales6–8.
El tratamiento es sintomático: fármacos antiepilépticos, tratamiento conductual, rehabilitación motora y tratamiento ortopédico en los casos de espasticidad.
En cuanto al pronóstico, desde el punto de vista cognitivo es muy malo. Las crisis se controlan mejor en la adolescencia y la edad adulta. Asimismo, la independencia motora puede mejorar conforme el niño crece9.
El objetivo de este trabajo es comparar nuestros hallazgos con los de otros autores y analizar las posibles correlaciones clinicogenéticas, identificando si existen diferencias destacables en los pacientes con estudio genético negativo, para así poder establecer un pronóstico evolutivo.
MATERIAL Y MÉTODOSEstudio descriptivo retrospectivo en el que se analizan las historias clínicas de los casos de SA diagnosticados en nuestro hospital y tratados en la Unidad de Neurología Pediátrica del Hospital Materno Infantil Carlos Haya de Málaga desde enero de 1981 hasta enero de 2007.
Se analizaron las características fenotípicas y conductuales, los resultados genéticos, el tipo de epilepsia, la edad de comienzo, las características electroencefalográficas (EEG), la respuesta al tratamiento antiepiléptico, las pruebas de neuroimagen y la evolución.
Para la evaluación del fenotipo, se utilizaron los criterios diagnósticos del consenso de SA de 20062. Para el diagnóstico genético, en todos los casos se efectuó un cariotipo estándar, análisis de metilación y bandeo FISH (hibridación fluorescente in situ). No se investigó la mutación del gen UB3A.
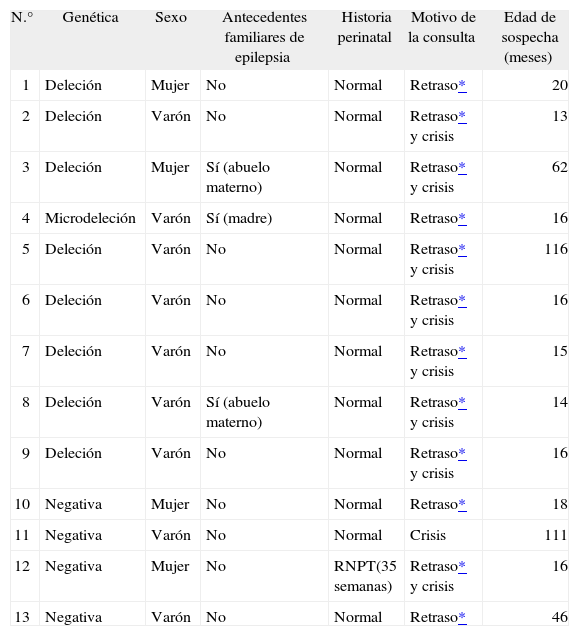
RESULTADOSSe incluyeron 13 casos, 9 con estudio genético positivo y 4 con genética negativa y fenotipo característico que cumplieron criterios clínicos. En las tablas 1 a 4 se exponen los datos principales.
Características generales
| N.° | Genética | Sexo | Antecedentes familiares de epilepsia | Historia perinatal | Motivo de la consulta | Edad de sospecha (meses) |
| 1 | Deleción | Mujer | No | Normal | Retraso* | 20 |
| 2 | Deleción | Varón | No | Normal | Retraso* y crisis | 13 |
| 3 | Deleción | Mujer | Sí (abuelo materno) | Normal | Retraso* y crisis | 62 |
| 4 | Microdeleción | Varón | Sí (madre) | Normal | Retraso* | 16 |
| 5 | Deleción | Varón | No | Normal | Retraso* y crisis | 116 |
| 6 | Deleción | Varón | No | Normal | Retraso* y crisis | 16 |
| 7 | Deleción | Varón | No | Normal | Retraso* y crisis | 15 |
| 8 | Deleción | Varón | Sí (abuelo materno) | Normal | Retraso* y crisis | 14 |
| 9 | Deleción | Varón | No | Normal | Retraso* y crisis | 16 |
| 10 | Negativa | Mujer | No | Normal | Retraso* | 18 |
| 11 | Negativa | Varón | No | Normal | Crisis | 111 |
| 12 | Negativa | Mujer | No | RNPT(35 semanas) | Retraso* y crisis | 16 |
| 13 | Negativa | Varón | No | Normal | Retraso* | 46 |
RNPT: recién nacido pretérmino.
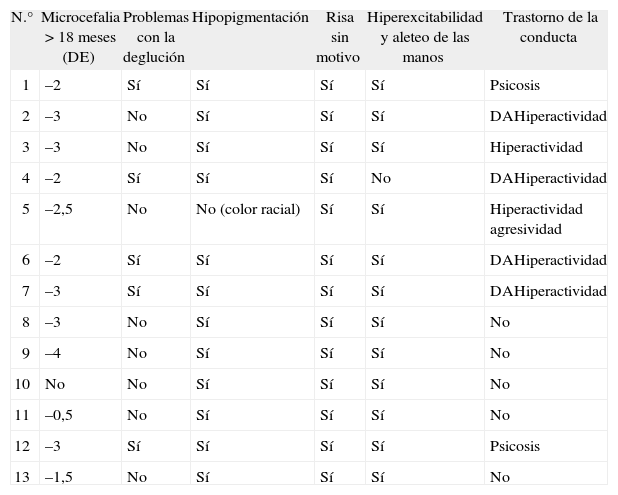
Fenotipo físico y conductual
| N.° | Microcefalia > 18 meses (DE) | Problemas con la deglución | Hipopigmentación | Risa sin motivo | Hiperexcitabilidad y aleteo de las manos | Trastorno de la conducta |
| 1 | –2 | Sí | Sí | Sí | Sí | Psicosis |
| 2 | –3 | No | Sí | Sí | Sí | DAHiperactividad |
| 3 | –3 | No | Sí | Sí | Sí | Hiperactividad |
| 4 | –2 | Sí | Sí | Sí | No | DAHiperactividad |
| 5 | –2,5 | No | No (color racial) | Sí | Sí | Hiperactividad agresividad |
| 6 | –2 | Sí | Sí | Sí | Sí | DAHiperactividad |
| 7 | –3 | Sí | Sí | Sí | Sí | DAHiperactividad |
| 8 | –3 | No | Sí | Sí | Sí | No |
| 9 | –4 | No | Sí | Sí | Sí | No |
| 10 | No | No | Sí | Sí | Sí | No |
| 11 | –0,5 | No | Sí | Sí | Sí | No |
| 12 | –3 | Sí | Sí | Sí | Sí | Psicosis |
| 13 | –1,5 | No | Sí | Sí | Sí | No |
DA: déficit de atención; DE: desviación estándar.
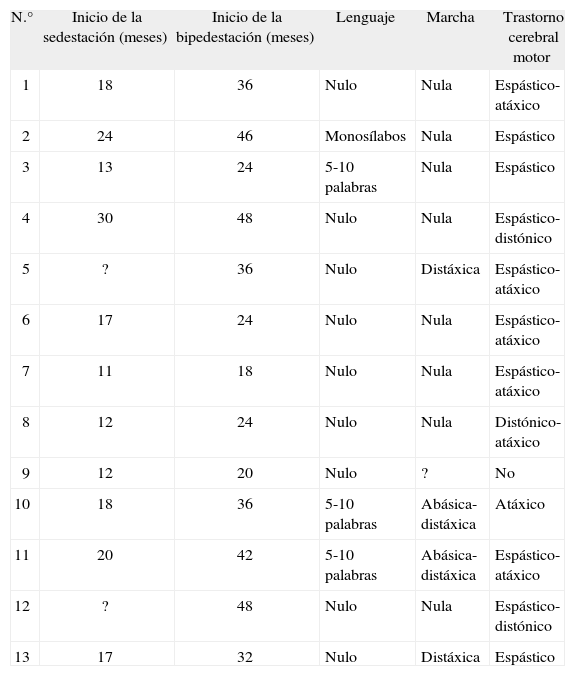
Desarrollo psicomotor
| N.° | Inicio de la sedestación (meses) | Inicio de la bipedestación (meses) | Lenguaje | Marcha | Trastorno cerebral motor |
| 1 | 18 | 36 | Nulo | Nula | Espástico-atáxico |
| 2 | 24 | 46 | Monosílabos | Nula | Espástico |
| 3 | 13 | 24 | 5-10 palabras | Nula | Espástico |
| 4 | 30 | 48 | Nulo | Nula | Espástico-distónico |
| 5 | ? | 36 | Nulo | Distáxica | Espástico-atáxico |
| 6 | 17 | 24 | Nulo | Nula | Espástico-atáxico |
| 7 | 11 | 18 | Nulo | Nula | Espástico-atáxico |
| 8 | 12 | 24 | Nulo | Nula | Distónico-atáxico |
| 9 | 12 | 20 | Nulo | ? | No |
| 10 | 18 | 36 | 5-10 palabras | Abásica-distáxica | Atáxico |
| 11 | 20 | 42 | 5-10 palabras | Abásica-distáxica | Espástico-atáxico |
| 12 | ? | 48 | Nulo | Nula | Espástico-distónico |
| 13 | 17 | 32 | Nulo | Distáxica | Espástico |
DA: déficit de atención; DE: desviación estándar.
Características de la epilepsia
| N.° | Edad de inicio (meses) | Crisis iniciales | Actividad de base característica | Localización de puntas o punta-onda | Tratamiento antiepiléptico actual |
| 1 | 17 | Mioclonías, espasmos* (hipsarritmia) | No inicialmente | Parietotemporal izquierda | AVP + CZP |
| 2 | 4 | Mioclonías | Sí | Occipital derecha | AVP + CZP |
| 3 | 24 | Ausencias, atónicas, parciales | Sí | Bioccipital | AVP |
| 4 | 7 | Mioclonías, ausencias | Sí | Temporal izquierda | AVP + CZP |
| 5 | 12 | Estado febril | Sí | Frontotemporal izquierda | AVP + CZP |
| 6 | 13 | Mioclonías, atónicas | Sí | Occipital izquierda | AVP + ESM |
| 7 | 17 | Mioclonías, ausencias | Sí | Temporal izquierda | AVP + LTG |
| 8 | 12 | Espasmos* (hipsarritmia en sueño) | No inicialmente | Generalizados | AVP + ESM |
| 9 | 16 | Mioclonías, atónicas, ausencias | Sí | Bioccipital, generalización | AVP + CZP |
| 10 | 9 | Atónicas | No inicialmente | Temporooccipital derecha | Control sin tratamiento |
| 11 | 3 | Mioclonías | Sí | - | AVP |
| 12 | 13 | Espasmos* | Sí | Frontal, generalización | AVP + Dexametasona |
| 13 | No crisis | No |
AVP: ácido valproico; CZP: clonazepam; ESM: etosuximida; LTG: lamotrigina.
Los 9 casos con confirmación genética correspondían a 8 casos por deleción y uno por microdeleción. Se observó un claro predominio del sexo masculino (69 %). No se registraron casos en hermanos. Únicamente 3 casos (23 %) refirieron antecedentes familiares de epilepsia, todos en la familia materna. La historia perinatal fue normal en todos los casos, salvo en el caso n.° 12, que se trató de un recién nacido pretérmino de 35 semanas que requirió ingreso por dificultad respiratoria de tipo II.
Las edades de diagnóstico estaban comprendidas entre los 13 y los 121 meses; la media fue de 37 meses (desviación estándar [DE]: 36,68) y la mediana fue de 16.
Las pruebas de laboratorio (hemograma, bioquímica y estudio metabólico básico) fueron normales en todos los casos. La resonancia magnética (RM) craneal demostró atrofia corticosubcortical leve en el 69 % de los casos.
Fenotipo físico (tabla 2)Once casos presentaron microcefalia a partir de los 18 meses, con una media entre –2 y –3 DE. La plagiocefalia fue prácticamente constante. En todos se evidenciaron alteraciones en la boca tipo prognatia, macrostomía, protusión lingual y dientes separados; pero sólo un 38 % de ellos presentaron problemas en la alimentación o la deglución. En 12 de ellos se constató la hipopigmentación de piel, ojos y cabellos (el caso n.o 5 era de origen sudamericano). Presentaron estrabismo 5 niños, 2 de ellos con nistagmo.
Fenotipo conductual (tabla 2)Todos los casos manifestaron risa sin motivo, característica que se hace más evidente con la edad. Un 92 % de los casos presentaron hiperexcitabilidad con hiperactividad y aleteo de manos. En 4 pacientes se evidenció apetencia por el agua e incremento de sensibilidad al calor. Las alteraciones del sueño se presentaron en 3 de 13 en forma de insomnio de conciliación y en 4 de 13 como despertares frecuentes. Uno de estos casos (n.° 8) se trató con melatonina con buena respuesta. Los trastornos de la conducta se constataron en un 58 %, y dos de ellos terminaron desarrollando conducta psicótica. De los casos con estudio genético negativo, salvo el caso n.o 12, que es de los que ha tenido peor evolución, los otros tres no presentaron trastornos de la conducta.
Desarrollo psicomotorLa edad media de inicio de la sedestación fue de 17,5 meses y la de la bipedestación, 33 meses. El tono muscular estuvo disminuido en todos los pacientes, con reflejos exaltados. Tanto el lenguaje como la marcha fueron las áreas más afectadas, tal y como se refleja en la tabla 3. En el 77 % de los casos se evidenció espasticidad. El control de esfínteres fue efectivo en 3 pacientes, 2 de ellos con estudio genético normal.
Características de epilepsia (tabla 4)Doce de los 13 casos presentaron crisis epilépticas. La edad media de inicio de las crisis fue de 12 meses (rango: 3-17), al igual que la mediana.
Las crisis iniciales más frecuentes fueron mioclonías (58 %), ausencias atípicas (33 %) y crisis atónicas (33 %). A lo largo de la evolución se observó una variación en el tipo de crisis, aumentando la frecuencia de mioclonías (12/12) clonías (5/12) y crisis parciales complejas (5/12). Se observaron con menor frecuencia estados febriles, parciales simples y versivas. No se constató ningún caso de crisis tónicas.
En cuanto a las características EEG, la actividad de base característica de ondas lentas hipervoltadas a 2-3Hz la presentaron inicialmente 10 de 12, y a lo largo de la evolución, 12 de 12. Se evidenció hipsarritmia en 2 casos, uno en vigilia y otro durante el sueño. La estimulación lumínica intermitente (ELI) fue positiva en el 58 % de los casos.
La asociación con paroxismos de puntas o puntas-ondas en el EEG intercrítico se observaron en todos los casos, excepto en uno. La localización más frecuente de estos paroxismos fue la occipital y la temporal. En los últimos EEG realizados a estos niños, la localización del paroxismo cambió en 4 casos con migración hacia zonas temporales; en un 58 % se evidenció indiferenciación.
En el único caso sin epilepsia, los primeros EEG fueron normales y posteriormente mostraron lentificación de actividad de base.
En cuanto al tratamiento, el fármaco inicial más empleado fue el ácido valproico (AVP) en 8 de 12, y a lo largo de la evolución, en 12 de 12. El segundo en frecuencia fue el clonazepam. Recibió corticoides (ACTH y dexametasona) una sola paciente (n.° 12), que además es la que ha estado con más fármacos al mismo tiempo (un total de cuatro). El control de las crisis se consiguió en el 92 % de los niños con tratamiento antiepiléptico; actualmente tan sólo hay un caso controlado sin tratamiento. Nueve de los 12 casos con epilepsia precisan dos fármacos para estar libres de crisis.
EvoluciónLa evolución fue desfavorable en todos los casos. Desde el punto de vista motor, un 54 % de ellos no tiene ninguna independencia motora. Desde el punto de vista cognitivo, en todos se constata retraso mental moderado-grave. Y en el desarrollo conductual, 4 pacientes mejoraron la sociabilidad, y en sólo 2 desapareció la hiperactividad.
DISCUSIÓNEl SA fue publicado por primera vez en 1965 por el pediatra Harry Angelman10. En 1989 se describe su asociación con la deleción del fragmento 11-13 del brazo largo del cromosoma 1511,12, y en 1997 se publica el papel del gen UB3A como causante del síndrome13; desde entonces se ha perfilado mucho su diagnóstico genético. Sin embargo, queda todavía un porcentaje considerable de casos con estudio genético negativo, a lo que hay que sumar que el estudio del gen UB3A no forma parte de la rutina diagnóstica de muchos laboratorios; por lo tanto, una buena orientación clínica es necesaria para solicitar un estudio genético guiado y específico, teniendo en cuenta que un resultado negativo no descarta el diagnóstico. Se necesita establecer protocolos de diagnóstico genético que sirvan de guía ante la sospecha clínica de SA, ya que es preciso conocer la causa genética para ofrecer un consejo genético, un pronóstico y un diagnóstico prenatal en posteriores embarazos.
Respecto a la correlación genotipo-fenotipo descrita, coincidimos en la presentación grave de los casos de deleciones. Tres de los casos con estudio genético normal presentan menos microcefalia y un mejor desarrollo psicomotor, sobre todo en la marcha. Diferimos en una de las niñas (n.o 12) que tienen estudio genético normal que ha tenido muy mala evolución motora, conductual y de las crisis. No sabemos si se debe a una mutación del gen o a otra causa aún no descrita. La hipopigmentación no sólo está presente en los casos de deleción. El único caso sin epilepsia y el único controlado sin tratamiento pertenecen al grupo con genética negativa. Son necesarios más casos para establecer relaciones fenotipos-genotipos consistentes.
La edad media de diagnóstico del SA ha descendido en los últimos años; esto sugiere un mejor conocimiento del síndrome y un grado mayor de sospecha diagnóstica. El diagnóstico precoz es fundamental para evitar pruebas complementarias innecesarias y para poder ofrecer una orientación temprana a los padres. Por debajo de 1 año es más difícil establecer un diagnóstico debido a la ausencia del fenotipo característico completo.
En cuanto a los problemas de alimentación, fundamentalmente en la deglución, la frecuencia (38 %) es más baja que en otras casuísticas publicadas; según las series, entre el 50 y el 77 %9,14. Esta discordancia podría deberse a que son síntomas que pueden pasar desapercibidos para los padres.
Las alteraciones del movimiento es uno de los síntomas más acentuados y constantes a lo largo de la evolución del niño. El aleteo de manos asociado a la hiperexcitabilidad es el más común, aunque no es exclusivo del SA, pues también se presenta en el síndrome del X frágil y el síndrome de Asperger14,15.
Los trastornos de sueño se atribuyen a un déficit de melatonina16. Su frecuencia global (53,8 %) es más baja que en otras series14,17.
El retraso en las primeras adquisiciones es considerable; la media de inicio de sedestación concuerda con otros registros, aunque la bipedestación es algo más precoz en nuestros datos14.
La epilepsia no es condición indispensable para el diagnóstico, sino que, tal y como se especifica en los criterios diagnósticos, está presente en el 80 % de los casos; en nuestra serie se constata en el 92 %. Esta elevada frecuencia podría deberse a que 8 de los 9 casos con estudio genético positivo son deleciones. La edad media de inicio de las crisis es más precoz que en otras publicaciones14,18,19. Destaca la prevalencia de mioclonías, que probablemente fueran de comienzo más precoz a lo referido, ya que con frecuencia pasan desapercibidas. La frecuencia de crisis mioclónicas coincide con la de otros registros, aunque se describe una gran variabilidad de crisis iniciales en el SA18. Como dato interesante llama la atención los 3 casos que inician el cuadro con espasmos en flexión; se trata de síndromes de West que genéticamente están condicionados a desarrollar un SA. La frecuencia del síndrome de West es algo superior que en otras casuísticas. Al igual que se refleja en la bibliografía, no encontramos crisis tónicas, una característica diferencial con el síndrome de Lenox-Gastaut. A lo largo del seguimiento encontramos EEG característico y sugestivo de SA en el 91 % de los casos con epilepsia, un porcentaje mayor de lo que muestran otras series18,19. El EEG puede ayudar a orientar el diagnóstico, sobre todo en aquellos casos con genética negativa. La localización de los paroxismos de puntas o puntas-ondas en zonas predominantemente posteriores se constata también en otras series20, y podría estar relacionado con la hipopigmentación de ojos de estos niños, que los hacen más sensibles a la luz. Asimismo, es una característica que facilita el diagnóstico diferencial con el síndrome de Rett, ya que en esta entidad los paroxismos de punta-onda se localizan con mayor frecuencia en las zonas anteriores.
En cuanto al tratamiento antiepiléptico, la combinación de AVP y clonazepam es la que ha demostrado ser más eficaz, tal y como se refleja en la bibliografía. Por otra parte, la carbamazepina y la vigabatrina pueden empeorar las crisis; este empeoramiento está relacionado con la anormalidad de los receptores GABA presentes en estos pacientes, ya que el gen GABRB3 está localizado en la región distal del 15q11-q1321–23.
La intensidad del grado de retraso motor y cognitivo hace necesario que cada niño con SA se someta a un programa de intervención y rehabilitación precoz e individualizado.
En conclusión, debido a que las características clínicas del SA son complejas y comunes, es fundamental conocer el síndrome y sus características fenotípicas para solicitar un estudio genético guiado y específico. El fenotipo característico que describió Angelman se comprueba en casi todos los pacientes.
Debido al número insuficiente de casos, no podemos establecer correlaciones significativas fenotipo-genotipo; sin embargo, nuestros resultados ponen de manifiesto la mayor gravedad y la peor evolución de los casos de deleciones.
A pesar de que no existe un tratamiento específico y conocemos su pronóstico, el interés que despierta su estudio se debe a las implicaciones que el retraso mental conlleva en el individuo afectado, en su familia y en la sociedad. Al establecer un diagnóstico específico, podemos explicar a los padres la causa, ofrecer un consejo genético e informarles sobre grupos de soporte y asociaciones; y al niño le evitaremos pruebas innecesarias, le brindaremos un tratamiento y una planificación educacional guiada.
