


Sr. Editor:
Presentamos este caso clínico en el que se ha detectado una microdeleción en el cromosoma 22, pudiendo ser ésta el mecanismo genético responsable de su expresión fenotípica.
Lactante de un mes de vida, de origen saharaui, que ingresa por presentar crisis tónico-clónicas generalizadas, estando afebril. Madre de 38 años, sufrió aborto previo, sin causa filiada.
En ecografías prenatales se detecta hemimelia derecha; no se realiza amniocentesis por deseo familiar. Parto a las 35 semanas. Peso nacimiento: 2.420 g. Posnatalmente se realizan ecocardiograma, ecografía vesicorrenal y cerebral (sin hallazgos patológicos) y radiografía de extremidad superior derecha.
Exploración física al ingreso peso 2.800 g (P < 3), talla 48,5 cm (P3), y perímetro craneal 36 cm (P25-50). Facies peculiar.
Analítica: hipocalcemia (calcio 5,8 mg/dl, calcio iónico 2,9 mg/dl), proteínas totales 6,1 g/dl, y albúmina 3,3 g/dl. Se descarta déficit de vitamina D (25[OH]D 11 ng/ml), hipomagnesemia (Mg de 1,4 mg/dl), ingesta elevada de fósforo (no constatada), causas yatrogénicas o alcalosis (pH 7,33 con bicarbonato de 21 mmol/l). El valor de PTH es bajo (16,1 ng/ml). En la radiografía de tórax se observa arco aórtico derecho y aplasia tímica (fig. 1), confirmadas en la TC torácica superior (fig. 2). Diagnóstico de hipocalcemia secundaria a hipoparatiroidismo congénito, incluido dentro del síndrome de DiGeorge. Cariotipo 46XX. FISH: microdeleción 22q11- (D22s75-).
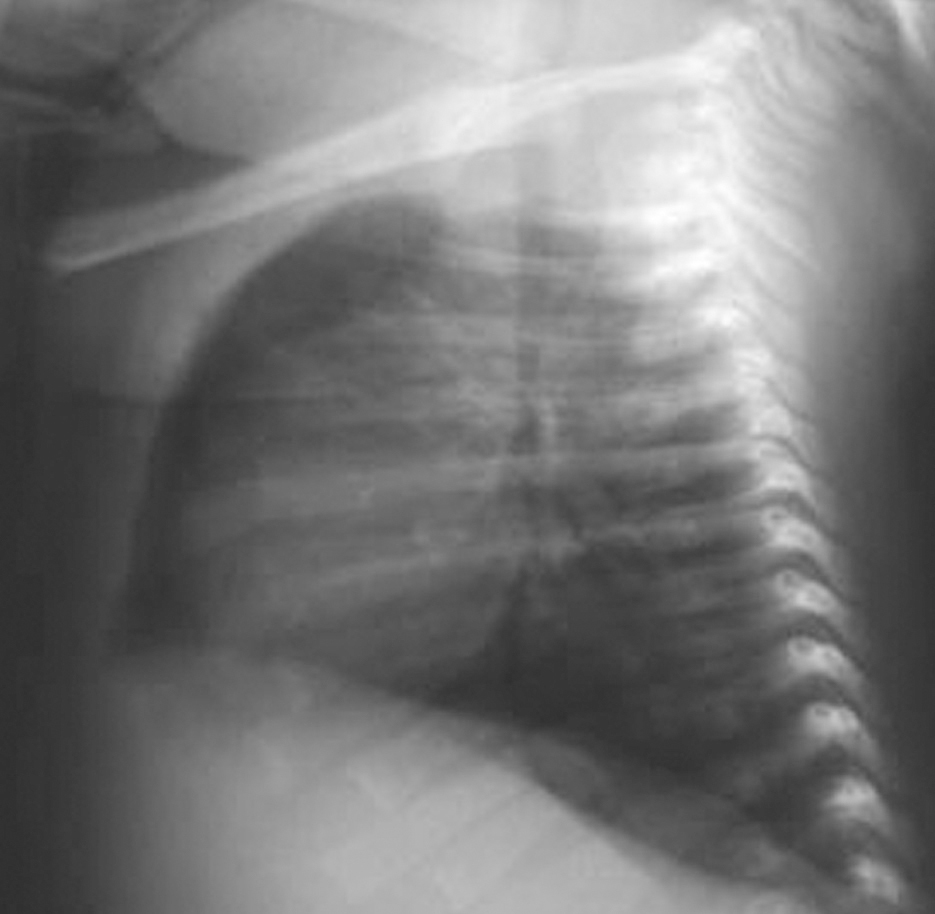
Figura 1.Radiografía lateral de tórax: ausencia de timoen el espacio retroesternal.
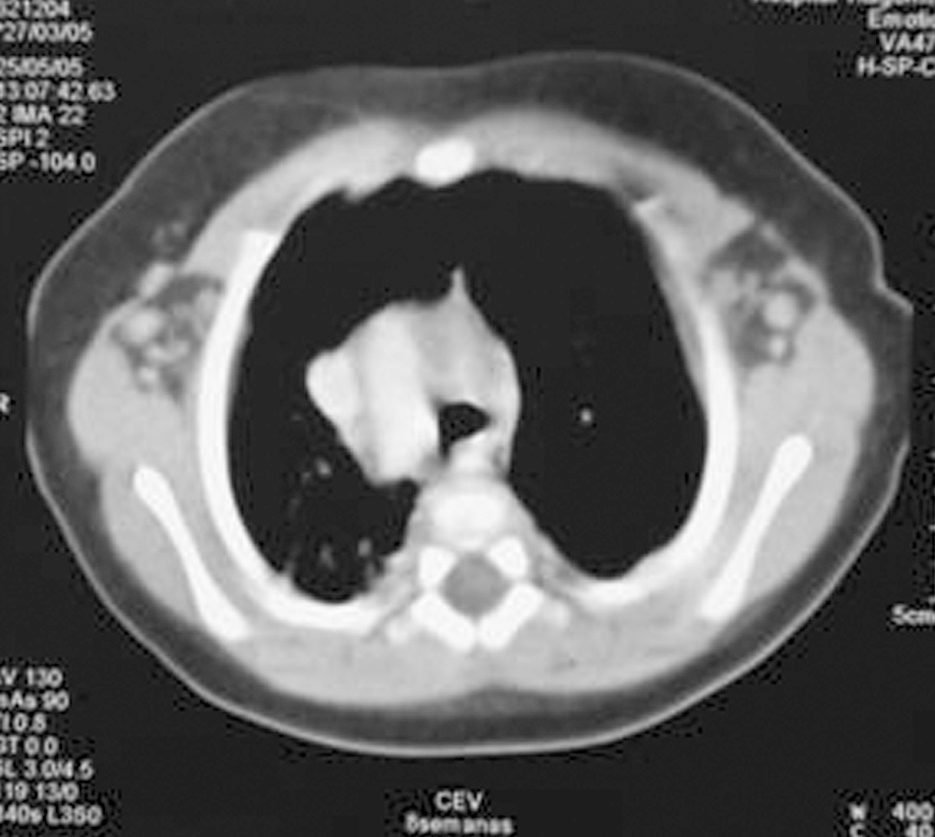
Figura 2.TC torácica superior: arco aórtico derecho. Ausencia de timo.
Se normaliza la calcemia mediante la administración de calcio por vía intravenosa, y posteriormente por vía oral, asociándose la administración de calcitriol.
No existen disfunciones inmunológicas. Actualmente la paciente es controlada en el Servicio de Endocrinología Pediátrica.
Es sabido que las malformaciones congénitas se producen durante la organogénesis, entre la tercera y la octava semanas de desarrollo. Es importante conocer los fenómenos que se producen durante cada etapa de la organogénesis: los esbozos de las extremidades se observan en forma de evaginaciones al final de la cuarta semana de gestación. En la regulación molecular del desarrollo de las extremidades tienen un papel determinante los genes HOX, también llamados genes de caja homeótica1 altamente conservados estructural y funcionalmente. Cambios sutiles en un segmento específico de una proteína HOX, pueden provocar fallos graves en el desarrollo2.
En la quinta semana, durante el desarrollo de la cabeza y el cuello del embrión, se forman los arcos, hendiduras y bolsas faríngeas, siendo el timo y las glándulas paratiroides derivados de la tercera y cuarta bolsas faríngeas. Parte del esqueleto facial y craneal, el estroma del tejido conjuntivo de las glándulas y también las almohadillas endocárdicas troncales derivan de células de la cresta neural que emigran hacia los arcos faríngeos.
El inicio clínico de nuestra paciente, consiste en crisis convulsivas debidas a la hipocalcemia secundaria a hipoparatiroidismo congénito, habiéndose descartado debidamente otras etiologías. El hipoparatiroidismo congénito puede deberse a mutaciones en los genes de la PTH, a mutaciones activas en el receptor sensor del calcio (CASR)3, a la agenesia de las glándulas paratiroides, a síndromes metabólicos, o al síndrome de DiGeorge. La ecografía cervical y TC torácica superior realizados revelan ausencia de timo y arco aórtico derecho asociado; todo ello sugestivo de síndrome de DiGeorge4.
Originalmente se postuló una etiología multifactorial del síndrome de DiGeorge. En 1981 de la Chapelle et al publicaron la primera asociación de síndrome de DiGeorge con un defecto cromosómico concreto subyacente, la microdeleción intersticial en la región 11 del brazo largo del cromosoma 225,6 cuya prevalencia estimada es 1/2.000 en la población general.
El cromosoma 22 sólo representa el 1,6-1,8 % del total del ADN genómico7 y la región q11 incluye 30 genes individuales. La microdeleción 22q11.2 afecta a la región donde se han identificado varios genes asociados a una variedad de fenotipos clínicos que incluye el síndrome de DiGeorge8,9. El caso descrito en nuestro trabajo, podría ser una variabilidad fenotípica de la microdeleción 22q11, que incluiría aplasia tímica, hipoparatiroidismo congénito, facies típica, anomalías de los grandes vasos y malformaciones de los miembros o por el contrario ser una asociación de malformaciones debidas a mecanismos genéticos diferentes.
Existe también un pequeño grupo de pacientes con síndrome de DiGeorge en quienes no se ha detectado esta microdeleción, sugiriendo asimismo heterogenicidad genética. Las mutaciones de genes HOX podrían tener alguna participación en la dismorfogénesis de las extremidades y derivados de la tercera y cuarta bolsas faríngeas. Por ello, cuando se establezca la correlación entre secuencias de ADN y bandas cromosómicas del cromosoma 22 y demás cromosomas, pasaremos a descifrar la estructura y funcionamiento de estos genes y su posible participación en los distintos síndromes dismorfológicos.
Correspondencia: Dra. E. Vera de Pedro.
Servicio de Pediatría. Hospital de Txagorritxu.
P. Vascongadas, 3, 4.º D. 01002 Vitoria. España.
Correo electrónico: elenaver2005@yahoo.es
