




El diagnóstico genético molecular permite la confirmación de numerosas enfermedades que anteriormente solo tenían un diagnóstico clínico. Por ello, es importante la configuración por el profesional médico de una base de datos, que salvaguardando la intimidad y cumpliendo la legislación vigente, permita la búsqueda de antiguos pacientes a los que ofrecer las posibilidades del diagnóstico genético molecular cuando el momento de ser diagnosticados clínicamente no había sido descubierta aún su base genética. El estudio molecular no solo permite la confirmación diagnóstica sino también el adecuado consejo genético, establecer el pronóstico e incluso realizar un eventual diagnóstico prenatal. A propósito de ello, referimos el caso ya publicado en 19901 de una paciente con síndrome de Ehlers-Danlos tipo I en la que se halla la mutación responsable 19 años después.
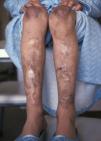
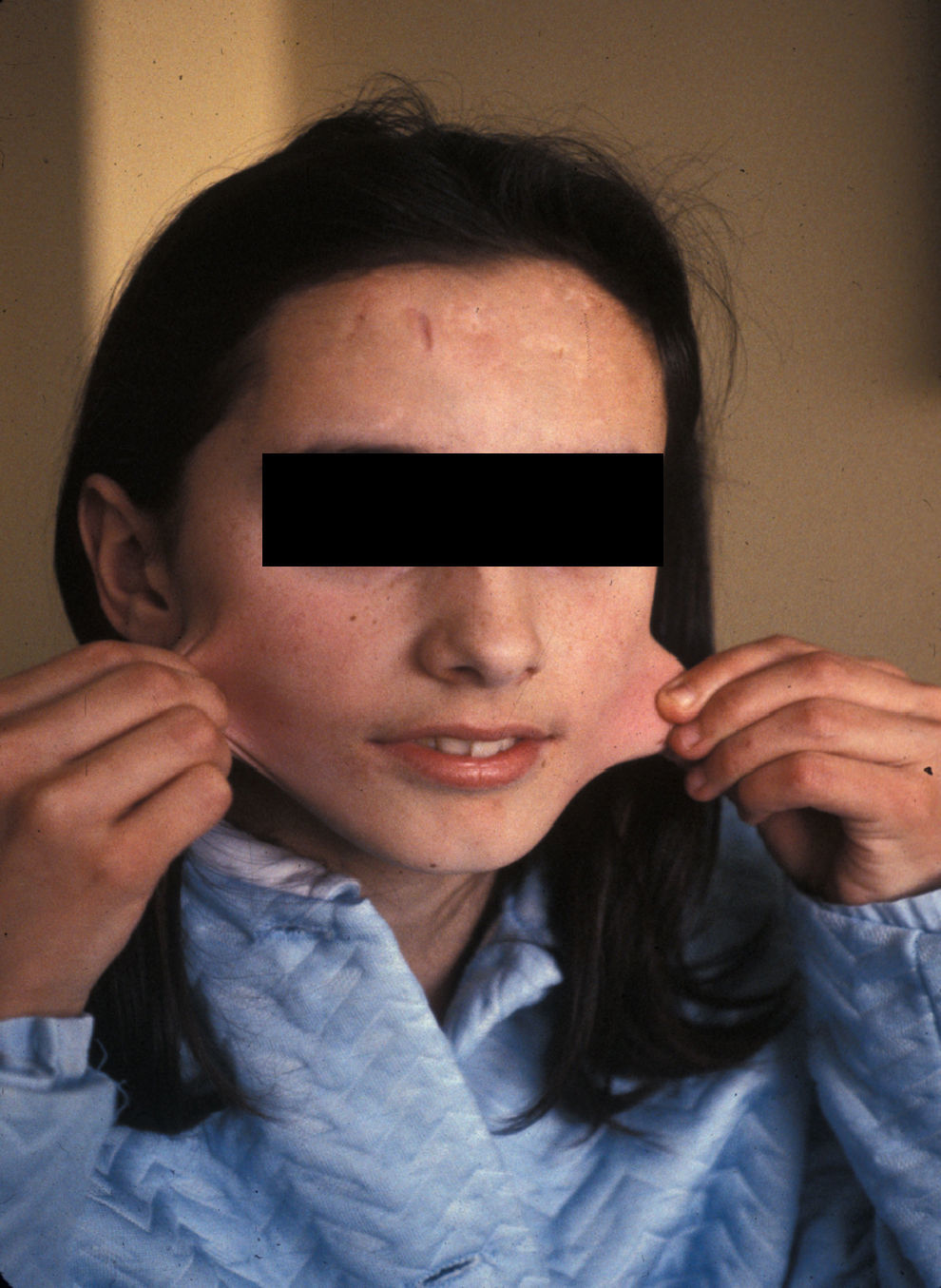
En el momento de la primera consulta, la paciente de 13 años presentaba cicatrices distróficas con aspecto de «papel de fumar» en frente, codos, rodillas y piernas (en relación con traumatismos banales típicos de la infancia), hiperextensibilidad cutánea e hipermovilidad articular (figs. 1 y 2), manifestadas desde la etapa de lactante. Sus padres, no consanguíneos, eran sanos al igual que su hermano menor. No se evidenciaron alteraciones oculares, dentales ni cardiológicas. En los estudios complementarios básicos se encontró como único hallazgo osteoporosis en tibias y peronés con metabolismo calcio/fósforo y vitamina D normales. La biopsia cutánea no mostró alteraciones relevantes.
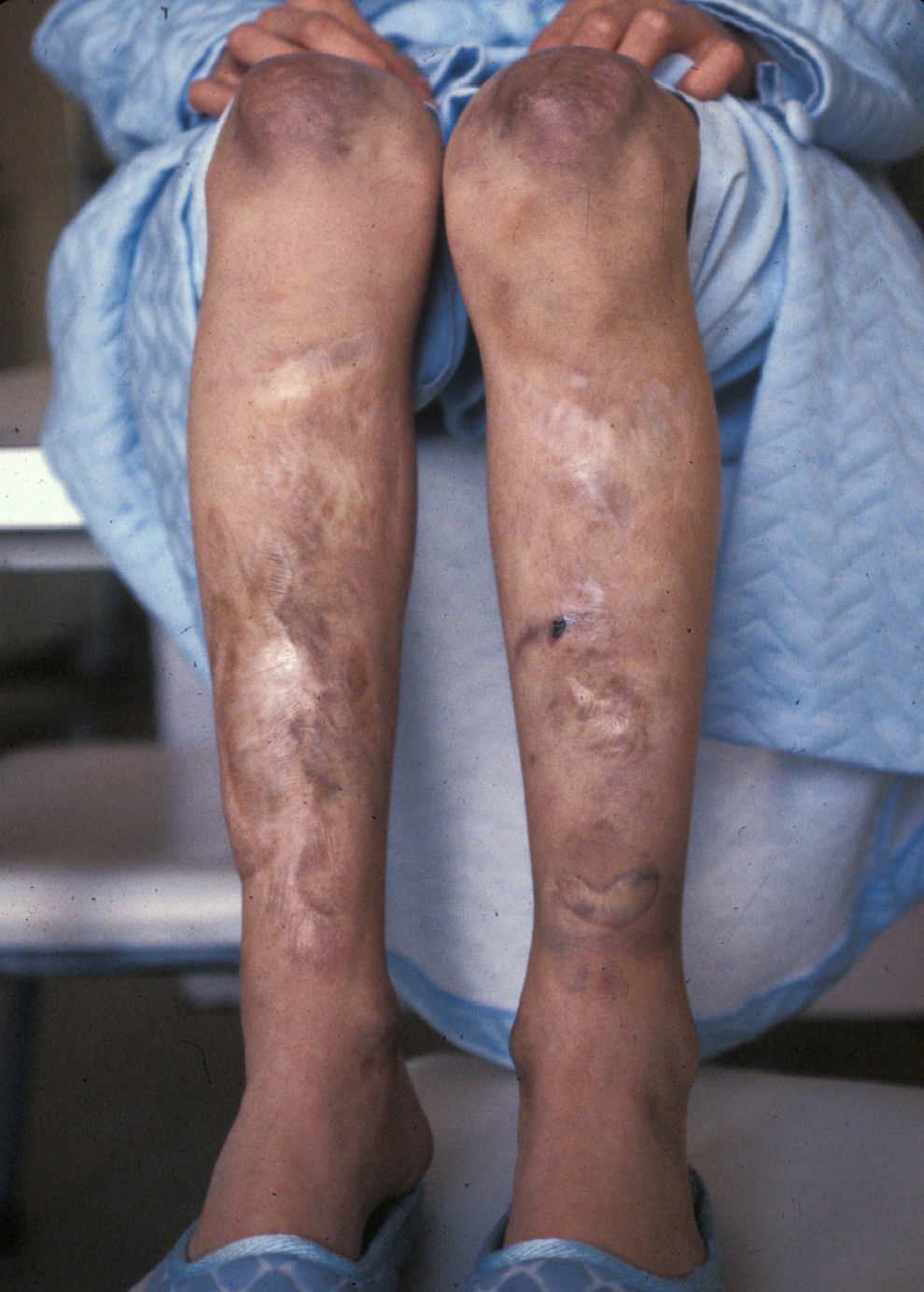
Ante la posibilidad de realizar estudios moleculares para el Síndrome de Ehlers-Danlos se contacta nuevamente con la paciente para ofrecerle el diagnóstico genético molecular y una eventual corrección de las lesiones dérmicas. En la actualidad (32 años) no presenta alteraciones funcionales sino las meramente estéticas derivadas de las cicatrices en codos y piernas. El estudio genético molecular detecta en heterocigosis la mutación «de novo» IVS19-1G>A en el gen COL5A1 que confirma el diagnóstico clínicamente establecido de síndrome de Ehlers-Danlos tipo clásico.
Esta colagenopatía comprende un grupo de alteraciones del tejido conectivo con distinta expresividad clínica2,3. Anteriormente se describían 8 subtipos4, pero en la actualidad se distinguen 6 tipos claramente definidos5: clásico (I y II), hipermóvil (III), vascular (IV), cifoescoliosis (VI), artrocalasia (VII) y dermatosparaxis (VII). Sus principales características son: hiperextensibilidad y fragilidad cutáneas, laxitud articular y facilidad para las equimosis. Cada subtipo presenta características fenotípicas específicas puesto que se relacionan con distintos genes que condicionan una alteración en diversas proteínas relacionadas con la estructura del colágeno (tabla 1)2,3,6,7.
Síndrome de Ehlers-Danlos: clasificación, clínica, herencia, defecto bioquímico y genética
| Tipo (Villefranche) | Antigua clasificación | Criterios diagnósticos mayores | Herencia | Alteración bioquímica | Gen y loci |
| Clásico | I (Gravis) |
| AD | Cadenas | COL5A1(9q34) |
| II (Mitis) | pro-α-1 y | COL5A2(2q14) | |||
| pro-α-2 de colágeno tipo V | |||||
| Hipermovilidad | III (Hipermovilidad benigna) |
| AD | Cadena | COL3A1(2q31) |
| pro-α-1 de colágeno tipo III | TNXB(6p21) | ||||
| Tenascina XB | |||||
| Vascular | IV (Arterial-equimótico) |
| AD | Cadena | COL3A1(2q31) |
| pro-α-1 de colágeno tipo III | |||||
| Cifoescoliosis | VI (Ocular-escoliosis) |
| AR | Lisilhidroxilasa | PLOD1(1p36) |
| Artrocalasia | VIIA y VIIB (Artrocalasia congénita múltiple) |
| AD | Cadenas pro-α-1 y pro-α-2 | COL1A1(17q21) |
| de colágeno tipo I | COL1A2(7q21) | ||||
| Dermatosparaxis | VIIC |
| AD | Peptidasa de procolágeno Lisiloxidasa | ADAMTS2 |
| (Dermatosparaxis humana) | (5q23) | ||||
| V | XR | ||||
| (Ligada a X) | |||||
| Otras formas | VIII | ? | |||
| (Periodontal) | AD | ? | (12p13) | ||
| X (Déficit en fibronectina) | ? |
El tipo I o gravis es la forma clásica severa de la enfermedad y se hereda con patrón autosómico dominante encontrándose el defecto genético en los genes que codifican para las cadenas pro-alpha-1 (V) y pro-alpha-2 (V) del colágeno tipo V (loci COL5A1 y COL5A2)8.
