



Recientemente hemos revisado en nuestro centro la experiencia en el tratamiento de las aplasias medulares adquiridas (AMA) y la citopenia refractaria de la infancia (CRI), siguiendo las recomendaciones y trabajos asociados a la European Society for Blood and Marrow Transplantation (EBMT)1,2. La AMA es la forma más común de insuficiencia medular adquirida, con una incidencia estimada de 1,5-2 millones de personas/año en Europa1. La CRI es el tipo más común de síndrome mielodisplásico en niños. En este grupo de pacientes se recomienda seguir la misma pauta de tratamiento establecido que en la AMA, basada en el trasplante de progenitores hematopoyéticos (TPH) o en la terapia de inmunosupresión (TIS)3. El objetivo de nuestro estudio fue evaluar de forma retrospectiva la tasa de respuesta, la supervivencia libre de evento y la supervivencia global de los pacientes con AMA y CRI sometidos a trasplante de progenitores hematopoyéticos o terapia inmunosupresora.
En este estudio se han incluido 48 pacientes tratados en el Hospital Infantil Universitario Niño Jesús, que han sido diagnosticados con AMA o CRI, entre los años 2001 y 2020. Dos pacientes (4,08%) no recibieron tratamiento, por recuperación espontánea el primero de ellos y por evolución como hemoglobinuria paroxística nocturna (HPN) con expansión del clon el segundo, sin precisar tratamiento hasta el momento de su traslado a otro centro. Definimos remisión completa (RC) alcanzar recuentos de ≥1,5×109/L neutrófilos, ≥150×109/L plaquetas y hemoglobina entre 10,5 y 13g/dL1. Se considera remisión parcial (RP) cuando el paciente alcanza independencia transfusional. Los eventos considerados para la supervivencia libre de enfermedad (SLE) son la recaída y el fallecimiento. Las curvas de incidencia acumulada se elaboraron con el estimador de supervivencia Kaplan-Meier, comparando sus resultados mediante log-rank.
La supervivencia global (SG) de la serie ha sido del 90,6%. Los 9 pacientes (18,75%) que se han sometido a TPH de familiar idéntico han alcanzado RC sin necesitar otra línea de tratamiento. En los 37 pacientes restantes (77,08%) se realizó TIS en primera línea con una tasa de respuesta del 73% (18/37 RC y 9/37 RP). Al comparar los resultados de la primera línea de tratamiento, la SG del TPH es del 100% frente al 85% para el TIS en primera línea (p=0,321). Sí resulta significativa la diferencia entre ambas modalidades de tratamiento al analizar la SLE: 100% para el TPH en primera línea frente al 47,1% para los sujetos tratados con TIS (p=0,038) (fig. 1A). La tasa de respuesta de los 13 pacientes que necesitaron un segundo TIS fue del 61,5% (4/13 RC y 4/13 RP). La SLE del TPH en segunda línea fue del 62,5% mientras que el TIS sólo alcanzó el 50%. De los 5 casos no respondedores al segundo ciclo TIS, 3 recibieron trasplante de donantes alternativos, permitiendo que la SG del TIS como segunda línea de tratamiento llegase al 83,9%. La mediana del intervalo de tiempo entre el diagnóstico hasta el inicio del TIS en primera línea ha sido de 23 días (0-180). Del estudio de los factores relacionados con la SG de estos pacientes se concluye que el tiempo que se tarda en comenzar el primer TIS influye negativamente en la SG de los pacientes. El retraso en el inicio del tratamiento más allá de 30 días desde el diagnóstico impacta de manera significativa en los resultados del tratamiento (96,3% vs. 48,1%) (p=0,005) (fig. 1B).
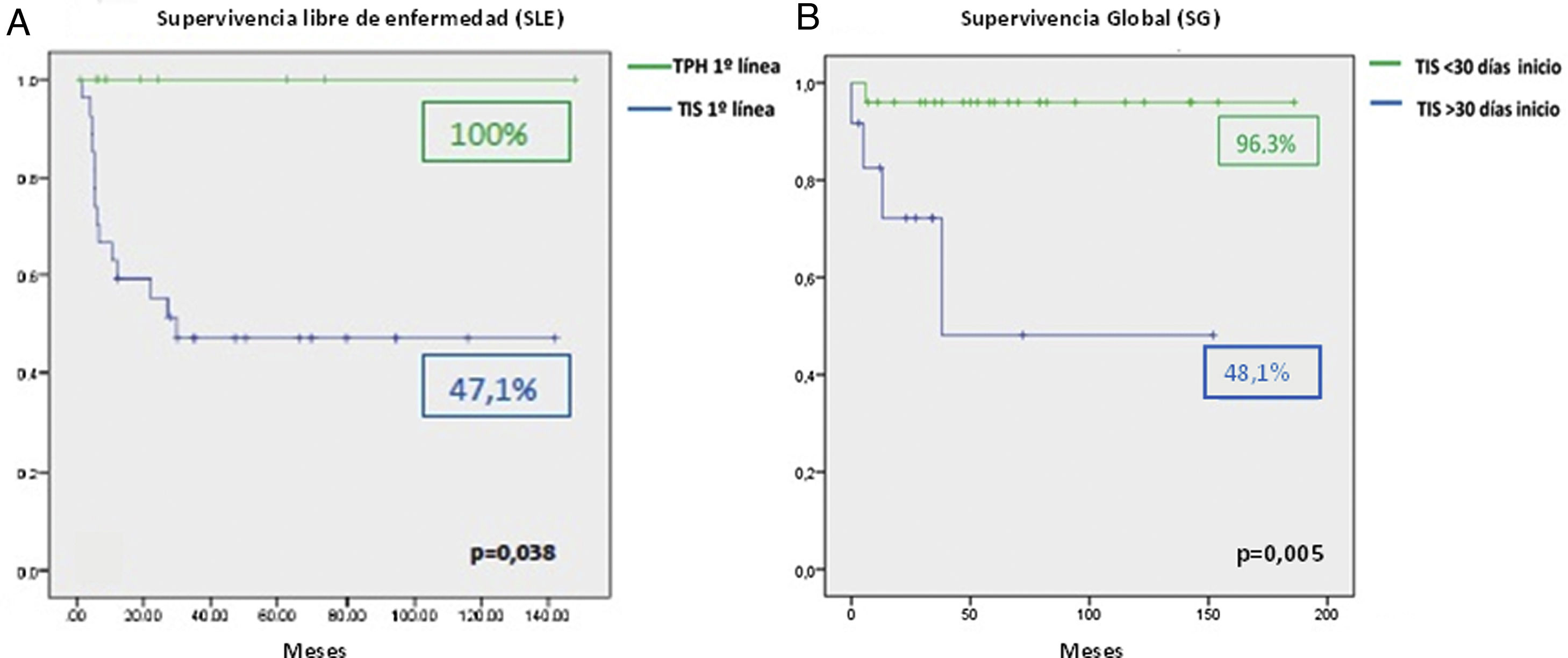
Los resultados obtenidos muestran que la SG de la primera línea de tratamiento se puede considerar óptima, siendo algo peores para la SLE. Estos resultados coinciden con los descritos en otras series4–6. Un tercio de los pacientes en nuestra serie no precisaron un trasplante alogénico y pudieron ser rescatados con TIS en ausencia de un donante familiar HLA idéntico. También se puede observar en nuestra serie que el TPH en segunda línea ofrece una buena opción de rescate tras un ciclo TIS con una supervivencia superior al 60%. Cabe destacar que los resultados entre grupos y líneas de tratamiento no deben ser interpretados para una mejor elección del tratamiento a realizar, ya que el carácter retrospectivo de nuestra serie solamente permite hacer una descripción de lo ocurrido con cada línea de tratamiento y no una comparación entre ellos (tabla 1).
Características de la cohorte total de pacientes y su tratamiento
| Número total de pacientes | 48 |
|---|---|
| Edad al diagnóstico (años) | 11 (8-14) |
| Mediana tiempo de seguimiento (meses) | 36 (13-79) |
| Género | |
| Masculino (%) | 33 (69%) |
| Femenino (%) | 15 (31%) |
| Diagnóstico | |
| Aplasia adquirida (%) | 33 (68,8%) |
| CRI (%) | 15 (31,3%) |
| TIS 1.alínea total (%) | 37 (77,08%) |
| ATGAM (%) | 21 (43,75%) |
| Timoglobulina (%) | 16 (33,33%) |
| Neutropenia grave | 21 (56,7%) |
| Dependencia transfusional | 24 (68,8%) |
| Remisión espontánea | 1 (2,08%) |
| Evolución a HPN | 1 (2,08%) |
| TPH total (%) | 20 (41,6%) |
| TPH 1.a línea (familiar HLA idéntico) (%) | 9 (18,75%) |
| TPH >1.a línea (%) | 11 (22,91%) |
| Acondicionamiento | |
| Mieloablativo | 5 (25%) |
| Toxicidad reducida | 15 (75%) |
| Evolución de pacientes con TPH | |
| Sin EICR | 14 (70%) |
| EICR agudo | 5 (25%) |
| EICR crónico | 1 (5%) |
| En remisión completa (%) | 16 (80%) |
| Exitus (%) | 4 (20%) |
| Evolución de pacientes con TIS | |
| En remisión completa (%) | 17 (35,4%) |
| Tratamiento activo con CSA (%) | 7 (18,9%) |
| Posterior TPH (%) | 11 (29,7%) |
| Exitus durante inmunosupresión (%) | 1 (2,7%) |
| Pérdida de seguimiento (%) | 1 (2,7%) |
Rangos expresados en cuartiles 25-75.
ATGAM: inmunoglobulina antilinfocitos T de origen equino; CMV: citomegalovirus humano; CRI: citopenia refractaria de la infancia; EICR: enfermedad injerto contra receptor; HLA: human leukocyte antigen; HPN: hemoglobinuria paroxística nocturna; TIS: tratamiento inmunosupresor; TPH: trasplante de progenitores hematopoyéticos.
En la actualidad se está estudiando la conveniencia de realizar el TPH a todos los pacientes en primera línea independientemente del tipo de donante debido a los excelentes resultados que se obtienen en este grupo de pacientes, como ocurre en nuestra serie cuando se recurre a donantes familiares idénticos. No obstante, nos parece de especial importancia resaltar que hasta un tercio de los pacientes pueden curarse exclusivamente con tratamiento inmunosupresor. Sin embargo, consideramos aún de mayor importancia destacar que, en caso de realizar TIS, el tratamiento debería iniciarse antes de los 30 días desde el diagnóstico para no perjudicar los resultados del mismo.
Presentación previa en reuniones LXIII Congreso Nacional SEHH - XXXVII Congreso Nacional SETH. Pamplona 2021.
