

Introducción
Los tumores neonatales se definen como los tumores que aparecen durante los primeros 28 días de vida y poseen unas características diferenciales respecto a los restantes tumores pediátricos 1. A pesar de su rareza (1,5-2 % de los tumores pediátricos) 2,3, se pueden encontrar los mismos tipos anatomopatológicos que en edades pediátricas posteriores, pero la inmadurez anatómica y fisiológica del período neonatal, condiciona un comportamiento especial que genera importantes dudas y enigmas respecto a su etiopatogenia, diagnóstico, pronóstico y tratamiento 4,5.
La incidencia de los tumores neonatales es difícil de estimar, por los diferentes conceptos aplicados por los autores para su selección y los escasos estudios publicados, que representan la experiencia de los principales hospitales pediátricos 6-27. Además, los tumores neonatales son clasificados en "benignos" o "malignos" basándose en criterios histológicos, inapropiados para el contexto global de estos tumores. El diagnóstico histológico de un tumor neonatal no es un indicador exacto de su comportamiento biológico y pronóstico evolutivo. Así, tumores benignos desde el punto de vista histológico pueden causar la muerte por su localización anatómica (p. ej., angiomas o teratomas cervicales). Por el contrario tumoraciones con histología maligna pueden presentar un comportamiento favorable, pudiendo incluso a regresar espontáneamente (p. ej., neuroblastoma). Por lo tanto, el seguimiento clínico-evolutivo es el que definirá la naturaleza benigna o maligna de la tumoración.
Según datos del Programa Surveillance Epidemiology and End Results (SEER) del National Cancer Institute's USA, la incidencia de cáncer en menores de 12 meses muestra un incremento anual, desde 183,4 por millón en 1970 a 189 en 1980 y 220 en 1990. De estos 220 casos, el 17 % fueron tumores neonatales, es decir 3,74 por 100.000 recién nacidos vivos/año. De los 7.000 niños diagnosticados de cáncer en EE.UU., el 10 % se diagnostican durante el primer año de vida, el 2 % durante el primer mes y sólo el 1 % en el primer día de vida. La prevalencia de neoplasias congénitas es de 1/12.500 a 13.700 total nacimientos 3. La distribución de las variedades histológicas en este registro (incluye tumoraciones histológicamente malignas más los nefromas mesoblásticos) muestra al neuroblastoma como el tumor neonatal más frecuente, seguido de las leucemias agudas, tumores renales, sarcomas, y tumores del sistema nervioso central (SNC). En los tumores de histología benigna el desconocimiento es aún mayor, pues la mayoría de las series publicadas se centran en las tumoraciones malignas. Los teratomas y los angiomas constituyen los tipos histológicos más frecuentes, aunque los datos son imprecisos y no existen cifras concretas de su incidencia y prevalencia 28,29.
Por la inmadurez anatómica y fisiológica del recién nacido, el tratamiento antineoplásico multidisciplinario, se adapta con objeto de evitar toxicidades y secuelas, teniendo en cuenta además las elevadas posibilidades de supervivencia de estos pacientes en centros especializados 30. El tratamiento más utilizado es el quirúrgico; sus limitaciones están condicionadas por la localización anatómica, ya que en algunas ocasiones se afectan órganos vitales 12,31,32. La quimioterapia está condicionada por las limitaciones fisiológicas del recién nacido que deben conocerse para elegir las dosis y vía de administración de los diversos fármacos quimioterápicos de forma adecuada. La absorción, biotransformación y excreción de estos fármacos, son diferentes a las de otras edades; así, la disminución del pH gástrico, los constantes cambios en las resistencias vasculares mesentéricas y periféricas, la inmadurez hepática y renal, la saturación de las proteínas plasmáticas, especialmente la albúmina, por la mayor afinidad y exceso de bilirrubina y ácidos grasos libres, el relativo aumento del volumen extracelular y los excesos de grasa en el recién nacido modifican la absorción, distribución, transporte, biodisponibilidad y eliminación de los agentes quimioterápicos. Para mantener la efectividad y minimizar la toxicidad, algunos autores y centros médicos especializados utilizan la mitad de la dosis habitualmente recomendadas en pediatría y la calculan en vez de por superficie corporal, por kilogramo de peso 12,33,34. La radioterapia suele omitirse por las graves secuelas a corto, medio y largo plazo 12,35.
Bader y Miller 36 cuando analizan la incidencia y mortalidad de los tumores neonatales, encuentran que ambos índices no se correlacionan. Globalmente, la tasa de mortalidad de las neoplasias neonatales, según el Third United States Cancer Survey 1, es una quinta parte de la incidencia, reflejando el buen pronóstico de las neoplasias del recién nacido. El Children's Hospital de Birmingham con una supervivencia del 55 % al año 20, y el St. Jude Children's Hospital del 68 % a los 11 años 10, constituyen las instituciones médicas de referencia.
El objetivo de nuestro estudio es analizar la frecuencia y las características clínicas, terapéuticas y evolutivas de los tumores neonatales en la última década, en un hospital infantil terciario.
Material y métodos
Estudio retrospectivo de las historias clínicas de los pacientes diagnosticados de tumor neonatal, atendidos en el Hospital Infantil Universitario La Fe de Valencia desde enero de 1990 a diciembre de 1999. Se realiza una descripción de las características anatómicas, histológicas, analíticas, clínicas, terapéuticas y evolutivas. Se excluyeron los hamartomas, hemangiomas cutáneos planos y cavernosos, nevos, lipomas, fibromas subcutáneos, linfangiomas y quistes epidérmicos. Las historias clínicas se obtuvieron a través del Servicio de Documentación y Archivo del Hospital Infantil Universitario La Fe, realizando una búsqueda según el diagnóstico de tumor y/o de sus diferentes tipos específicos.
Se realiza una revisión bibliográfica sistemática de los últimos 25 años sobre los tumores neonatales obtenida del Medline, Cancerlit, Index Citation Science y Embase. El perfil de búsqueda utilizado fue la combinación de "neonatal, congenital" y "tumors, cancer, neoplasms".
Resultados
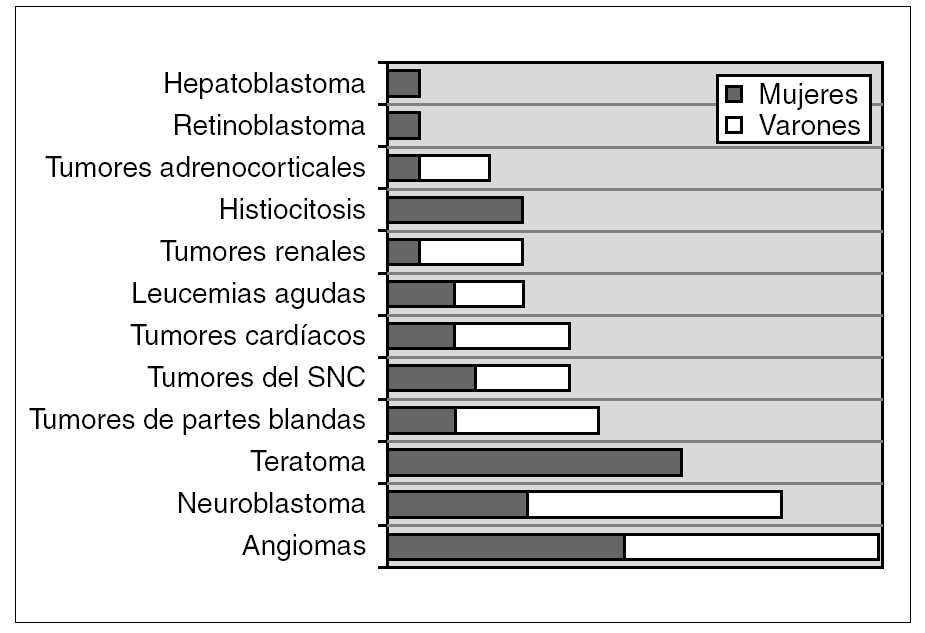
Se identificaron 72 tumores neonatales, que corresponden al 2,8 % de los tumores pediátricos diagnosticados en nuestro hospital durante dicha década. El hemangioma ha sido la tumoración más frecuente (20,8 %, 15 casos), seguido por el neuroblastoma (16,7 %, 12 casos), teratoma (12,5 %, 9 casos) y tumor de partes blandas (9,7 %, 7 casos). La distribución por variedades histológicas y género se representa en la figura 1. Globalmente, hay un ligero predominio de varones con 40 casos (relación 1,2:1).
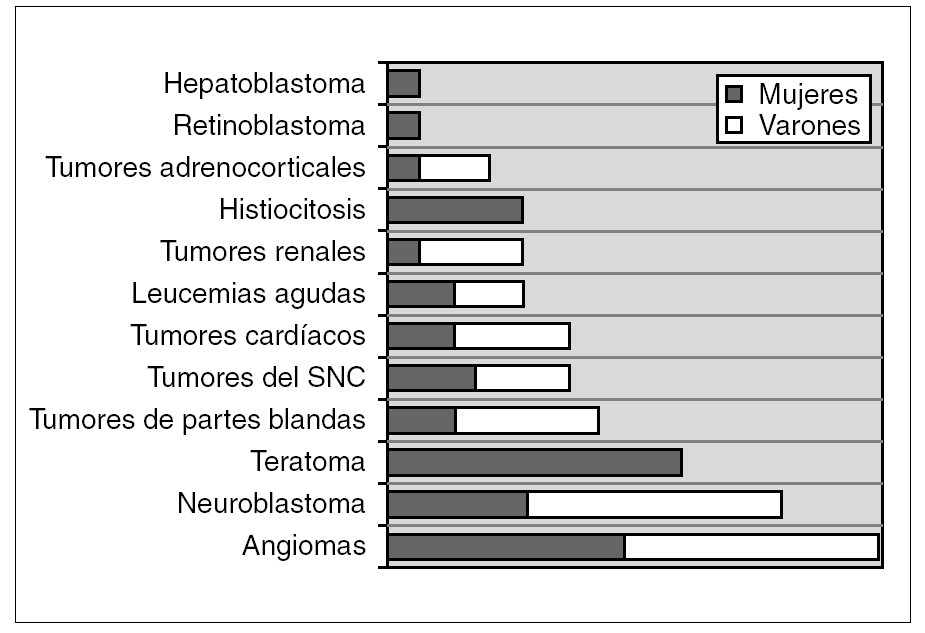
Figura 1. Distribución de los diferentes grupos tumorales y género (n.º casos).
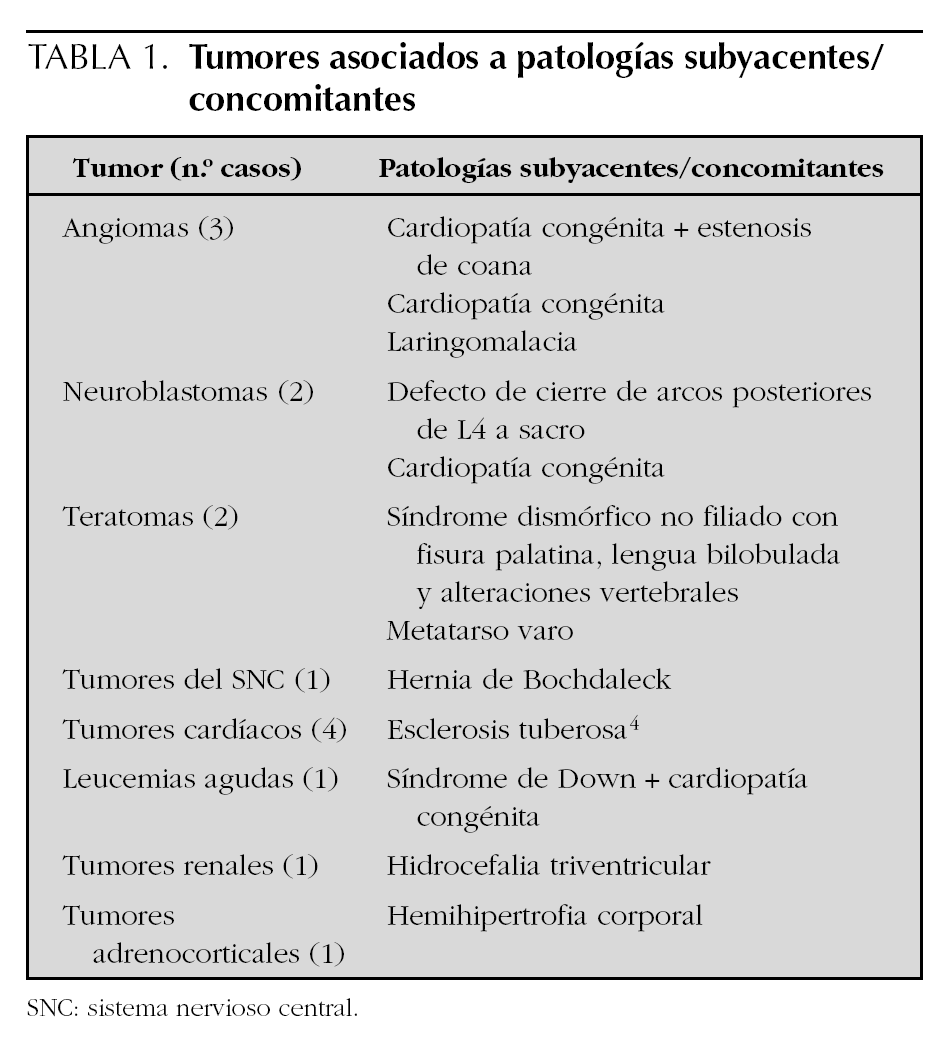
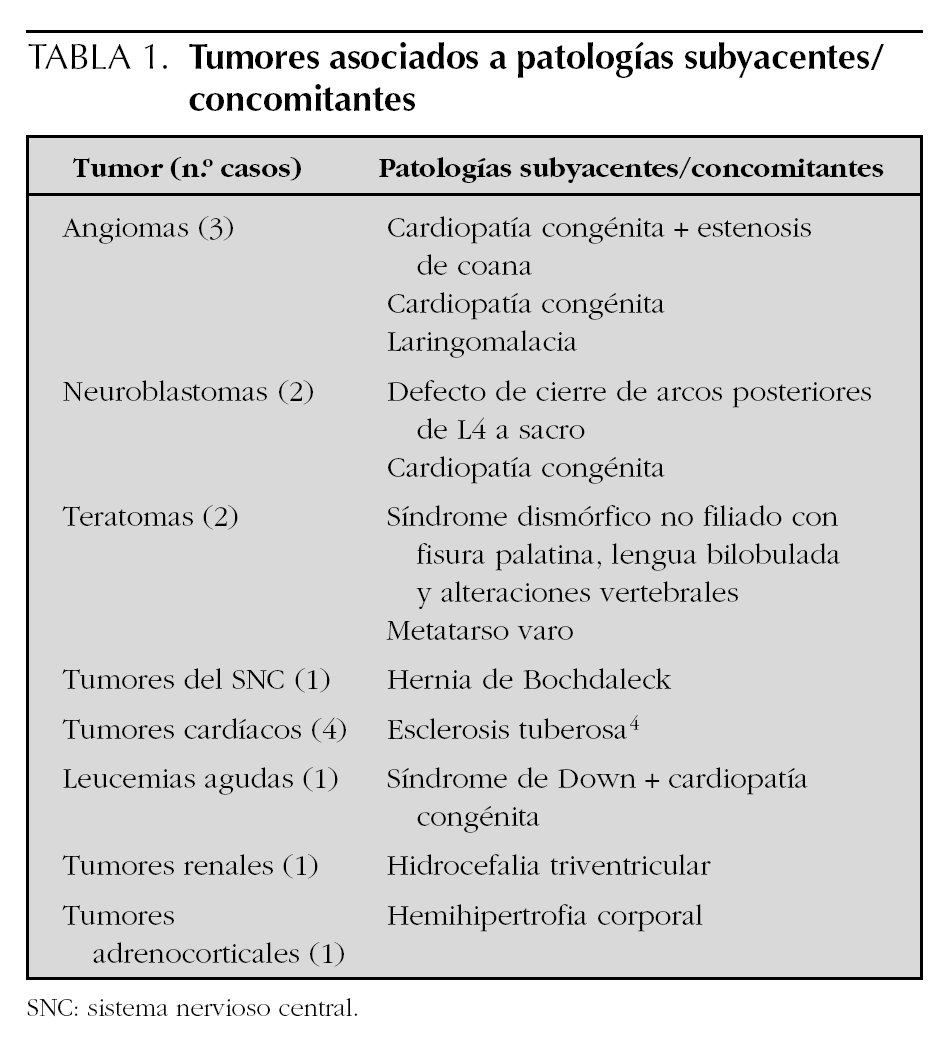
Mientras que el inicio de las manifestaciones clínicas fue al nacimiento en más de la mitad de los casos (41 casos), en 16 casos el diagnóstico fue prenatal. En el 86 % de los casos (62 casos) el diagnóstico se realizó durante la primera semana de vida. La sintomatología predominante fue la visualización de una tumoración o lesión cutánea en 24 casos (33,3 %), la presencia de una masa abdominal o hepatomegalia en 13 casos (18 %) y alteraciones respiratorias en 9 casos (12,5 %). Otras manifestaciones clínicas menos frecuentes fueron: vómitos, hidrocefalia, estrabismo y shock hemorrágico. Sólo en 5 pacientes el hallazgo de la tumoración fue casual. En 15 casos (20,8 %) existía alguna malformación, enfermedad o síndrome asociado (tabla 1). Las cardiopatías congénitas y la esclerosis tuberosa fueron las más frecuentes.
Respecto al tratamiento, la cirugía ha sido la opción más empleada, aplicándose en el 50 % de nuestros pacientes (36 casos). En 10 casos (13,9 %) se administró quimioterapia en monoterapia o con carácter coadyuvante. Otros tratamientos aplicados con menor frecuencia fueron la radioterapia (3 casos), interferón (3 casos), trasplante de progenitores hematopoyéticos (2 casos) y láser CO2 (3 casos). No se administró tratamiento en 19 pacientes (26,4 %).
En 11 pacientes se documentó la regresión espontánea de la tumoración, en seis de forma completa (dos neuroblastomas, dos histiocitosis y dos miofibromatosis) y en cinco parcial (un neuroblastoma mediastínico, un rabdomioma cardíaco y tres angiomas hepáticos).
Fallecieron 20 pacientes (27,8 %). El mayor número de fallecidos corresponde al grupo con tumores del SNC (5 casos), seguido de las leucemias agudas (3 casos) y los tumores cardíacos (3 casos). La mayor mortalidad por grupos histológicos, excluyendo al hepatoblastoma y retinoblastoma, que fallecieron, la registraron las leucemias (75 %) y los tumores del SNC (83,3 %), seguidos de los pacientes con histiocitosis y tumores cardíacos (50 %). Por el contrario, ninguno de los pacientes diagnosticados con hemangiomas y teratomas falleció, destacando así mismo la escasa mortalidad de los neuroblastomas (8,3 %). La supervivencia a fecha de enero 2005 es del 73 %, con una mediana de seguimiento de 8 años (rango: 0-15 años). De los supervivientes, tres presentan una afectación neurológica grave (dos tumores cardíacos con esclerosis tuberosa y el tumor cerebral) y dos malformaciones corporales (hemihipertrofia en tumor adrenocortical y escoliosis en miofibromatosis).
A continuación se comentan los resultados específicos de cada grupo de tumores.
Angiomas
Con 15 casos constituye la variedad histológica más frecuente de nuestra serie. Nueve asocian angiomas cutáneos. Ninguno de los pacientes falleció, aunque en la mayoría de los casos persisten las lesiones. Los aspectos clínicos, terapéuticos y evolutivos figuran en la tabla 2.

Neuroblastoma
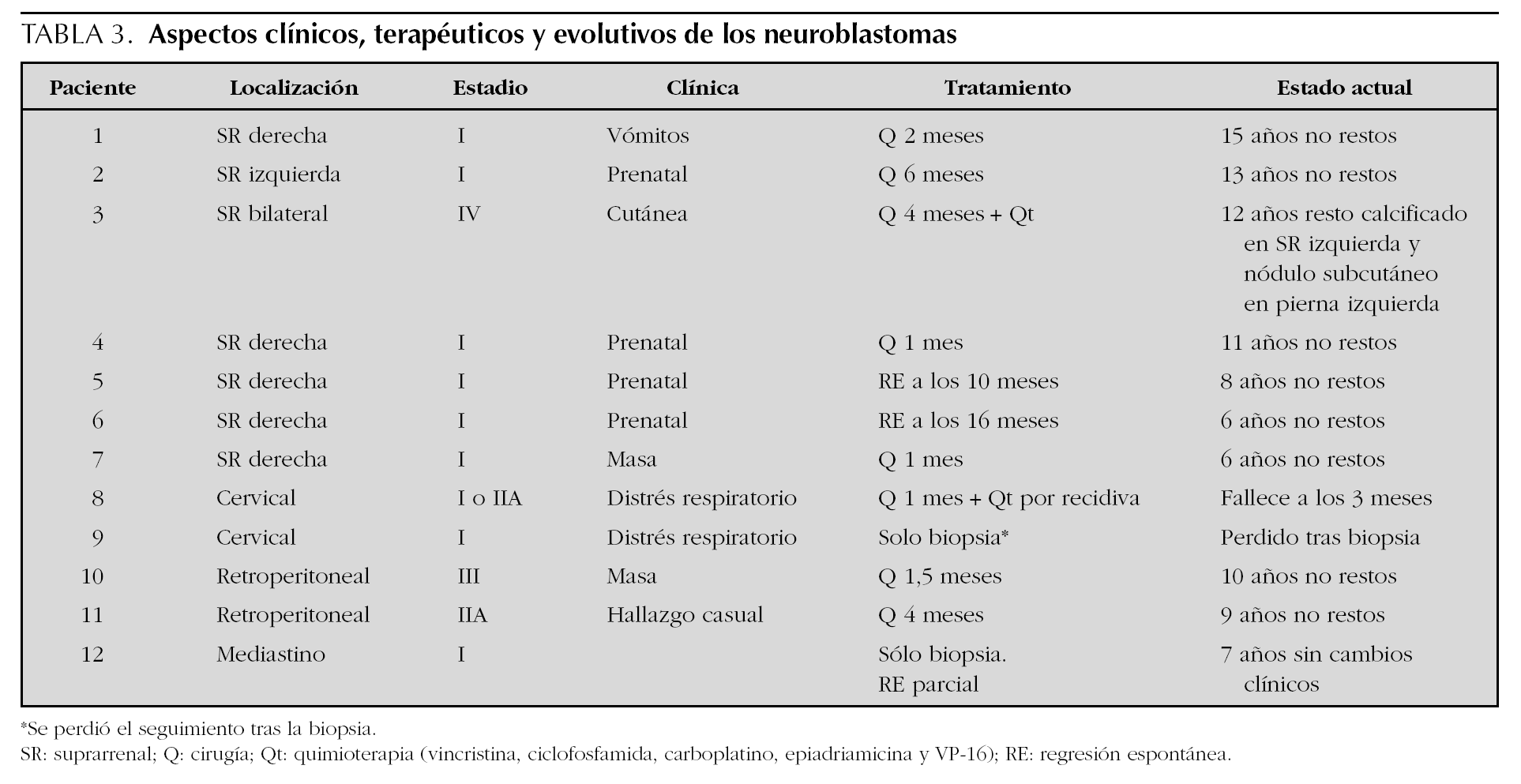
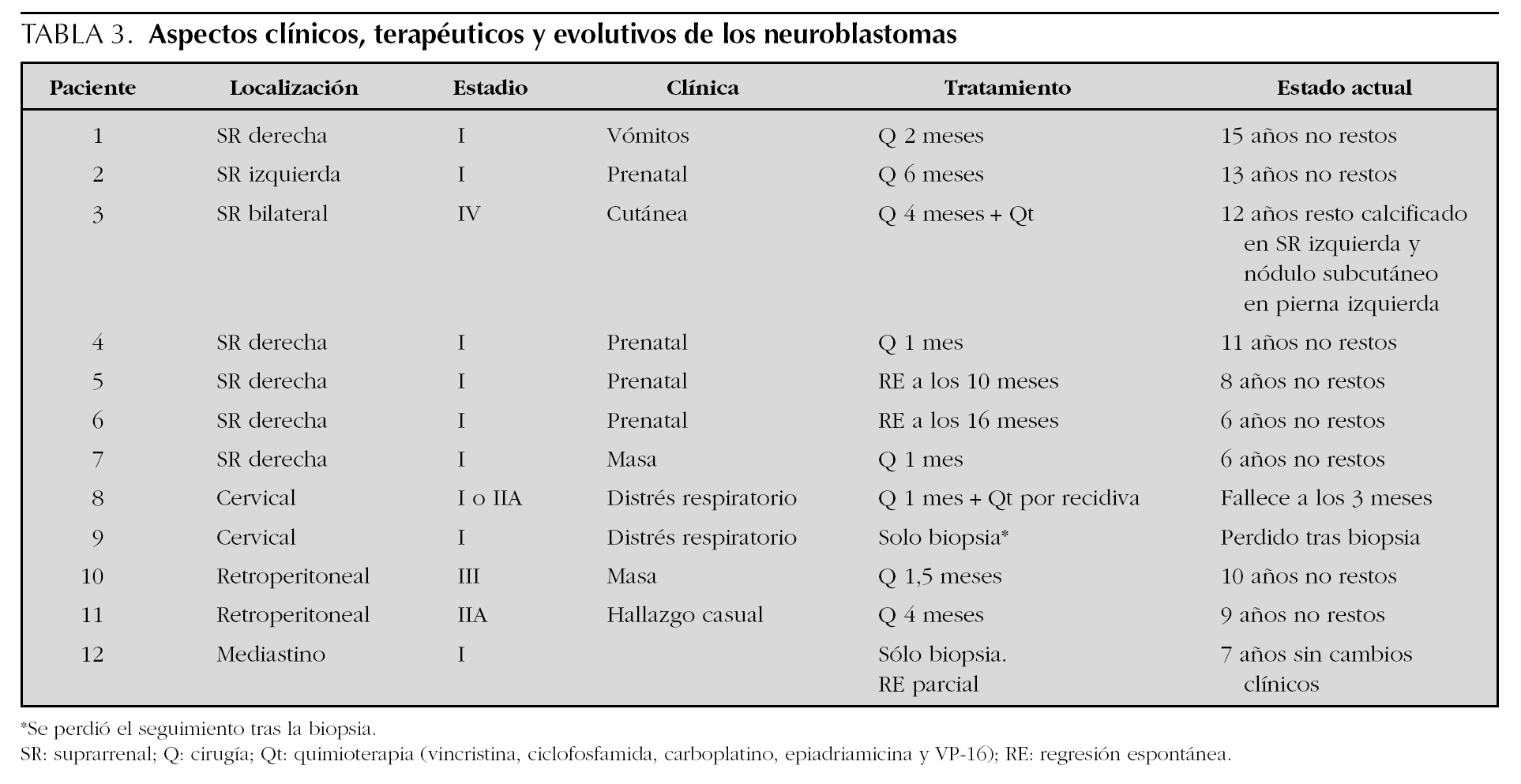
Con 12 casos, ocupa el segundo lugar en frecuencia tras los angiomas. Los aspectos clínicos, terapéuticos y evolutivos figuran en la tabla 3. En todos los pacientes se realizó una ecografía y tomografía computarizada de la zona afectada, completándose el estudio de extensión mediante la realización de una gammagrafía con 123I-MIBG (metaiodobencilguanidina) y punción de médula ósea en el 75 y 58 % de los casos, respectivamente. Todos los casos presentaron valores normales de los metabolitos urinarios de las catecolaminas y las cifras de enolasa neuronal específica y ferritina estaban elevadas en 6 y 10 casos, respectivamente. En 10 pacientes se obtuvo muestra histológica tumoral (8 resecados y 2 biopsiados) y en seis se realizó estudio citogenético. Ninguno de ellos presentaba amplificación del N-myc ni deleción 1p. El tratamiento quirúrgico se aplicó en 8 pacientes, asociándose en dos quimioterapia. En los restantes 4 pacientes no se aplicó tratamiento. La supervivencia se sitúa en el 91,6 % (11/12) tras una mediana de seguimiento de 9 años. Sólo un paciente falleció a los 3 meses de vida en el contexto de un cuadro séptico bajo el tratamiento quimioterápico instaurado por recidiva tumoral.
Teratomas
Si excluimos los teratomas de localización cardíaca (un caso) y cerebral (un caso), cuyo comportamiento clínico es determinado por su localización anatómica y no por su histología, registramos 9 teratomas. Todos sin signos histológicos de malignidad. La localización más frecuente fue sacrococcígea en 5 casos, siendo los restantes a nivel cervical, abdominal, mediastínico y orofaríngeo. La forma de presentación fue por diagnóstico prenatal entre la semana 25 y 33 en 4 pacientes y la detección de una tumoración al nacer en los cinco restantes. Dos pacientes fueron prematuros y no asociaron polihidramnios. El tratamiento fue quirúrgico, con resección completa en todos los casos. Tres pacientes presentaron complicaciones posquirúrgicas (paresia diafragmática izquierda en el teratoma mediastínico, ascitis quilosa y sepsis en el teratoma abdominal y epifisiólisis traumática femoral en uno de los teratomas sacrococcígeos), con resolución posterior. En 4 de 6 pacientes cuya resección se realizó tras la primera semana de vida, se analizó la alfafetoproteína plasmática, con cifras patológicas en todos ellos. Todos son supervivientes y sin secuelas.
Tumores de partes blandas
Se registraron 7 tumores de partes blandas: cuatro fibrosarcomas, un rabdomiosarcoma y dos miofibromatosis. Fibrosarcomas: tres se presentaron en extremidades (dos en muslo y uno en rodilla), y uno en la zona retroauricular. Recibieron tratamiento quirúrgico los 4 pacientes, consiguiéndose la curación en tres de ellos. El paciente con afectación retroauricular precisó quimioterapia y radioterapia paliativa por recidiva ganglionar a los 6 meses de edad. Falleció a los 10 meses de edad bajo tratamiento paliativo. Rabdomiosarcoma: se diagnosticó en una niña con múltiples tumoraciones cutáneas al nacimiento en cuello, axila, tórax, rodillas y frontal. Con diagnóstico clínico de angiomatosis múltiple, recibió tratamiento con prednisona, falleciendo a los 2 meses de vida por progresión tumoral. El diagnóstico necrópsico fue de rabdomiosarcoma embrionario en la región frontal con metástasis múltiples (pulmonar, mediastínica, retroperitoneal, mesentérica, subcutánea y renal). Miofibromatosis: se objetivó al nacimiento una tumoración paravertebral dorsal en los 2 casos. Tras la biopsia se realizó un seguimiento evolutivo, sin aplicarse tratamiento y regresando espontáneamente entre los 10 y 24 meses. Un paciente presenta escoliosis secundaria a su tumoración.
Tumores del SNC
Se registraron 6 tumores del SNC: 3 tumores neuroectodérmicos primitivos, un ganglioglioma, un astrocitoma y un teratoma. El diagnóstico prenatal se realizó en 3 pacientes, por aumento de diámetro biparietal, ventriculomegalia o hidrocefalia. Otras manifestaciones fueron abombamiento de la fontanela anterior, tumoración facial y exoftalmos. El tratamiento fue quirúrgico en 5 casos (resección subtotal), recibiendo quimioterapia 2 casos, uno como única terapéutica (carboplatino y etopósido, seguido de 6 ciclos de epirrubicina, vincristina, actinomicina D, ifosfamida y etopósido). De los 6 pacientes, cinco fallecieron (83,3 %), tres durante la intervención quirúrgica, uno en el postoperatorio por hemorragia intracraneal, y el restante, pasado el período neonatal por recidiva y progresión tumoral. El único superviviente fue diagnosticado de ganglioglioma y presenta un resto tumoral estable tras resección subtotal pero con importantes secuelas: retraso psicomotor, hidrocefalia con válvula de derivación ventrículo-peritoneal, epilepsia y sordera neurosensorial.
Tumores cardíacos
Se registraron 6 tumores cardíacos. Cinco rabdomiomas (dos con diagnóstico anatomopatológico [uno por biopsia del tumor y en el otro en la necropsia], y tres por la presentación y evolución clínica) y un teratoma (post mortem en la necropsia del recién nacido). Dos rabdomiomas eran multicéntricos. Dos casos fueron diagnosticados prenatalmente, y los restantes durante el primer día de vida por sospecha de cardiopatía congénita. La presencia de un hidrops fue la manifestación inicial en un caso. De los 5 rabdomiomas, cuatro fueron diagnosticados de esclerosis tuberosa (tres entre los 3 y 18 meses de vida, y uno en la necropsia). El teratoma no presentaba malformaciones asociadas. Ninguno de los pacientes recibió tratamiento. De los 6 pacientes, tres fallecieron por parada cardiorrespiratoria y arritmia cardíaca (taquicardia supraventricular). Dos de los 3 supervivientes, con esclerosis tuberosa, presentan un importante deterioro neurológico. Un paciente desarrolló un astrocitoma subependimario que motivó la
necesidad de una derivación ventrículo-peritoneal. En uno de ellos las múltiples tumoraciones cardíacas han regresado quedando sólo una en el ventrículo derecho y otra en el izquierdo.
Leucemias agudas
Se registraron 4 leucemias agudas, una de ellas en un paciente con síndrome de Down. Tres eran mieloides (LMA), con 2 casos de subtipo M5 y uno M7. El paciente restante presentaba una leucemia linfoide con morfología L2 de la clasificación FAB. La forma de presentación fue variable: palidez y hepatoesplenomegalia, lesiones cutáneas azuladas generalizadas con posterior infiltración testicular, y en 2 casos fue un hallazgo casual objetivado en el hemograma al ingreso por patología respiratoria (distrés respiratorio transitorio), asociando uno de ellos hepatomegalia. El estudio citogenético se realizó en 3 pacientes y los resultados fueron los siguientes: 46XX normal, en el paciente con LMA M7; 47XY + 21, en uno de los pacientes con LMA M5; y 47XY t 10,19 (q22;p13), + I (der 10), en el otro paciente con LMA M5. Los 4 pacientes recibieron quimioterapia con ARA-C, VM 26, prednisona, vincristina, L-asparraginasa, epiadriamicina, VP-16 y ciclofosfamida. A dos de ellos se les realizó trasplante de progenitores hematopoyéticos, y en un paciente se aplicó radioterapia por recidiva meníngea a los 5 meses de edad. Tres pacientes fallecieron entre los 6 y 17 meses de vida, pero sólo dos por progresión de su enfermedad, falleciendo el tercero por su cardiopatía congénita asociada (síndrome de Down). El único superviviente (LMA M5) tras el tratamiento citostático y trasplante de progenitores hematopoyéticos a los 9 meses de vida, permanece libre de enfermedad a los 9 años de edad.
Tumores renales
Se registraron 4 tumores renales, todos nefromas mesoblásticos. La forma de presentación más frecuente fue la masa abdominal (incluido uno con diagnóstico prenatal). Dos pacientes fueron prematuros, uno de ellos con polihidramnios. Un paciente asociaba una malformación neurológica congénita (hidrocefalia triventricular). Un paciente desarrolló hipertensión arterial y otro hipercalcemia, ambas de carácter transitorio, que se resolvieron tras la nefrectomía. Todos los casos fueron tratados con nefrourecterectomía. Hubo un fallecimiento en el postoperatorio por un fallo multiorgánico secundario a un proceso séptico (presentaba la malformación neurológica). Uno de los tumores evidenció mayor agresividad local, infiltrando estructuras extrarrenales (colon descendente y bazo) y precisó colectomía segmentaria con colostomía y esplenectomía. La anatomía patológica demostró un patrón celular y bordes quirúrgicos con infiltración tumoral. A pesar de no recibir quimioterapia, no ha presentado ninguna recidiva local tras 15 años de seguimiento.
Histiocitosis
Se registraron 4 casos. Dos con afectación orgánica múltiple, uno con afectación cutánea y lesión ósea única y otro con afectación exclusivamente cutánea. Aunque la forma de presentación fue la aparición de un exantema al nacimiento, las formas diseminadas se acompañaban de hepatomegalia en un caso y anemia en el otro. El diagnóstico se realizó por punción de médula ósea en las formas diseminadas y biopsia cutánea en las restantes. Evolutivamente, las dos formas diseminadas fallecieron, una por una sepsis fúngica, y la otra por progresión de su enfermedad. Este caso recibió quimioterapia con etopósido, prednisona y vinblastina. Las otras dos formas se resolvieron espontáneamente entre los 6 y 4 meses de vida.
Tumores adrenocorticales
Se registraron un carcinoma adrenocortical y dos citomegalias adrenocorticales. El paciente con carcinoma adrenal presentó shock hipovolémico por hemorragia suprarrenal que condicionó una suprarrenalectomía derecha al segundo día de vida. Desarrolló fenotipo Cushing a los 47 días de vida, observándose una masa suprarrenal derecha que se resecó. Las determinaciones hormonales mostraron un cortisol elevado y supresión de hormona adrenocorticotropa (ACTH). A los 10 días de la cirugía, inició recidiva local con gran deterioro clínico iniciándose mitotane. Falleció por progresión de su enfermedad a los 6 meses de edad. Los 2 pacientes con citomegalia adrenocortical fueron diagnosticados prenatalmente. Al nacimiento, la exploración física fue normal en uno de ellos y detectándose una masa en el hipocondrio derecho en el otro. Ante la posibilidad de neuroblastoma se realizó determinación de ferritina, enolasa neuronal específica y catecolaminas en orina, siendo normales. Se realizó resección completa tumoral mediante suprarrenalectomía que determinó el diagnóstico definitivo. Uno de los pacientes presenta una hemihipertrofía corporal derecha.
Retinoblastoma
Se registró un retinoblastoma bilateral con antecedentes familiares oftalmológicos: padre con retinitis pigmentaria y madre diagnosticada a los 14 meses de vida de retinoblastoma bilateral. Desarrolló estrabismo convergente del ojo derecho desde la primera semana de vida. Tras diagnóstico y estudio de extensión, precisó crioterapia del ojo derecho, y radioterapia externa del ojo izquierdo a los 2 meses de edad, con evolución satisfactoria. A los 4 años desarrolló rabdomiosarcoma alveolar en la región temporal izquierda, que tras remisiones parciales con cirugía y quimioterapia, falleció a los 6 años de edad por progresión tumoral.
Hepatoblastomas
De los 6 tumores hepáticos, cinco fueron angiomas y uno hepatoblastoma. El hepatoblastoma se presentó con gran hepatomegalia, distensión abdominal y circulación colateral. Las técnicas de imagen (ecografía y tomografía axial) demostraron lesión a nivel del segmento posterior del lóbulo derecho, heterogénea, de predominio hipoecoico, mal delimitada, de 5 * 4 * 4 cm, con vascularización aumentada y sin afectación de órganos extrahepáticos. Con diagnóstico de hemangioendotelioma, recibió tratamiento con prednisona e interferón, presentando progresivamente insuficiencia cardíaca y pulmonar, secundarias al crecimiento tumoral. Se realizó hepatectomía a los 6 meses de vida, falleciendo en el acto quirúrgico. El diagnóstico fue de hepatoblastoma epitelial y embrionario. Las determinaciones de alfafetoproteína eran patológicas desde el nacimiento, asociando hipercolesterolemia e hipertrigliceridemia desde los primeros días de vida. Los 5 angiomas se comentaron en el apartado correspondiente.
Discusión
Los tumores neonatales poseen una serie de características diferenciales respecto a los originados en épocas posteriores, constituyendo un subgrupo especial de tumores pediátricos. Para designarlos la bibliografía utiliza diferentes términos (tumores congénitos, tumores fetales, tumores perinatales, tumores del bebé o niño pequeño, embriomas o tumores embrionarios y tumores neonatales o del recién nacido) que son empleados como sinónimos cuando en realidad definen a subgrupos diferentes. El término más aceptado es el de "tumor neonatal" que incluye exclusivamente los tumores de los primeros 28 días de vida 1. Éste ha sido el criterio utilizado en nuestro trabajo de revisión.
Los casos registrados en nuestra serie suponen el 2,8 % de los tumores atendidos en nuestro hospital durante la década de 1990. Es ligeramente superior al obtenido en otras publicaciones, que los sitúan en el 2 % 2,3, y posiblemente se explique por los diferentes criterios de inclusión. La distribución de las diferentes variedades tumorales es similar a nuestras series anteriores, predominando las tumoraciones sólidas sobre las hematológicas. Según nuestros datos, la presente revisión constituye la mayor casuística publicada en nuestro país y una de las más amplias a nivel mundial 6-27. Los hemangiomas, neuroblastomas, teratomas y tumores de partes blandas son las variedades de tumor neonatal más frecuentes.
La incidencia de los tumores neonatales es imprecisa y de difícil estimación debido en parte a los diferentes conceptos aplicados por los autores para su definición y a los escasos estudios publicados, que representan la experiencia de los principales hospitales pediátricos con criterios de selección variables 6-27. Los criterios anatomopatológicos de malignidad/benignidad son relativos, siendo el seguimiento clínico-evolutivo el que definirá la naturaleza benigna o maligna de la tumoración. De esta forma, los estudios de tumores con histología maligna sitúan la incidencia de cáncer neonatal entre el 1,7 y 3,74 por 100.000 recién nacidos vivos/año 3,6,7,36. Por el contrario, los trabajos de tumores con histología benigna son escasos y sin datos numéricos concretos en su incidencia y prevalencia 28,29. Un estudio basado en la casuística del Children's Hospital de Birmingham 20 reveló una incidencia de 7,2 por 100.000 recién nacidos vivos/año. Incluyó a todas las tumoraciones tanto malignas como benignas y el criterio de selección abarcó hasta los 3 meses de edad, y aunque no es exclusivo del período neonatal, es representativo de todos ellos. De acuerdo con este estudio, la edad del diagnóstico, sexo e histología, eran similares a los de trabajos previos y objetivaron un incremento en la incidencia anual durante dicho período, pasando del 4,3 entre 1960-1969 al 10,4 por 100.000 recién nacidos vivos/año entre 1980-1989. El incremento en la incidencia de los tumores neonatales observado en éste y otros estudios, es debido principalmente al aumento de las tumoraciones sólidas, teratomas y neuroblastomas, permaneciendo constantes los tumores del SNC y leucemias agudas. Posiblemente, este incremento sea debido al avance y uso rutinario de técnicas de diagnóstico por imagen durante el embarazo, como es la ecografía fetal 3,37,38, así como a la introducción en algunos países europeos, Japón y Canadá del cribado de catecolaminas para el diagnóstico precoz del neuroblastoma 39-44. Con respecto a los datos publicados por nuestro hospital en años anteriores 28,29,45,46, objetivamos que el número de casos por año ha aumentado, pasando de 3,8 tumores por año en el período de 1971-1992 a casi el doble, 7,2 tumores por año en el período de 1990-1999. Independientemente de la posible influencia de los factores etiopatogénicos, atribuimos este incremento a un mejor y mayor uso (ecografías fetales y neonatales) de las pruebas diagnósticas, y a la selección más exhaustiva de pacientes realizada en la década de estudio.
El inicio de las manifestaciones clínicas (86 % antes de la primera semana de vida), la sintomatología predominante (visualización de una tumoración o lesión cutánea y masa abdominal o hepatomegalia) y las diferentes modalidades terapéuticas (cirugía la más empleada, seguido de quimioterapia en monoterapia o con carácter coadyuvante), son similares tanto al estudio realizado con anterioridad en nuestro hospital 28,29,45,46 como a las series publicadas por otras instituciones médicas 6-27. Las dosis y vía de administración de las diversas drogas quimioterápicas aplicadas se realizaron teniendo en cuenta las características fisiológicas del recién nacido en relación a la inmadurez en los mecanismos de absorción, biotransformación y excreción de estos fármacos y así mantener la efectividad y minimizar la toxicidad. Sólo en 3 pacientes seleccionados se aplicó radioterapia (fibrosarcoma retroauricular, leucemia linfoblástica y retinoblastoma) y en todos ellos fuera del período neonatal, hecho similar a otras publicaciones en las que la radioterapia suele limitarse u omitirse por las graves secuelas a corto, medio y largo plazo 12,35.
En 15 casos (20,8 %) existía alguna malformación, enfermedad o síndrome asociado, siendo las cardiopatías congénitas y la esclerosis tuberosa las más frecuentes. La asociación entre anomalías congénitas y tumores está bien establecida, con frecuencias inferiores a nuestra serie (15 %) 14,47-50. Estudios en animales de experimentación y en autopsias fetales humanas, sugieren que determinados factores potencialmente tóxicos, mutagénicos y cancerígenos transplacentarios originan efectos diferentes según la fase fetal evolutiva de actuación 51,52. Así, producirán abortos en la primera semana de vida fetal, malformaciones entre la segunda y la octava, y tumores entre la sexta y la 40 semanas de gestación. El período en que se inicia un tumor abarca desde el final de la organogénesis y se extiende durante toda la histogénesis. Así se explican los casos coexistentes de malformaciones congénitas y tumores, en los que la actuación de un mismo agente entre la sexta y la octava semana de vida fetal desencadena el desarrollo asociado de una malformación y un tumor.
La supervivencia en nuestra serie, en el momento del estudio (enero 2005), se sitúa en el 73 %, con una mediana de seguimiento de 8 años, siendo inferior al 85 % obtenido en el período previo en nuestro hospital 28,29,45,46. No obstante, es superior a la del Children's Hospital de Birmingham 20 y el St. Jude Children's Hospital 10, dos de las instituciones médicas con mayor supervivencia descrita, del 55 y 68 %, respectivamente. Estas diferencias posiblemente se expliquen por los diferentes criterios de inclusión, tanto en el período de estudio previo como en las series internacionales.
La etiopatogenia de los tumores neonatales es diferente a la encontrada en otras épocas de la vida 1,51,53-58. Generalmente, los tumores aparecen durante la segunda mitad de la vida, porque los factores cancerígenos (físicos, químicos o biológicos) precisan de largos períodos de latencia para producir las mutaciones, en los protooncogenes y en los genes supresores tumorales, y lógicamente, en la edad neonatal, este período es corto. Las principales vías de la oncogénesis implicadas en los tumores neonatales, son la preconcepcional y la transplacentaria, en las que los factores ambientales pueden actuar sobre las células germinales de los progenitores (oncogénesis preconcepcional) 51,59 o sobre el feto durante las 40 semanas de gestación (oncogénesis transplacentaria) 52,60-62. Un tipo especial de oncogénesis preconcepcional es la transgeneracional que originará los cánceres hereditarios o familiares en la que la mutación se hereda con carácter recesivo o dominante 63,64. Debido al corto período neonatal, podemos asumir que en los tumores neonatales no hay tiempo para actuar la oncogénesis posnatal.
El mecanismo de oncogénesis preconcepcional o transplacentario es originado por diversos factores ambientales recogidos en múltiples y diversos estudios epidemiológicos que han objetivado un incremento de cánceres en descendientes de progenitores expuestos preconcepcional o transplacentariamente a radiaciones ionizantes, no ionizantes, agentes infecciosos, fármacos y drogas, tabaco, alcohol, dieta y ocupaciones parentales, maternas y paternas, pero todo ello sobre una base genética o constitucional 55. Aunque no es objetivo de este trabajo, muchos de estos factores de riesgo ambientales están presentes en los casos estudiados y serán comentados en una publicación posterior.
Otra vía etiopatogénica de adquisición del cáncer neonatal y propia de este período de la vida es la transmisión materno-fetal de células tumorales, no de mutaciones. Aunque la placenta actúa como una barrera activa, física e inmunológica, condicionando un mínimo riesgo de transferencia de células tumorales, se han descrito metástasis materno-fetales de algunas neoplasias (melanomas, coriocarcinomas, linfomas, carcinomas broncogénicos y epiteliomas mamarios) 65-67. Su extraordinaria rareza ratifica que en nuestra reducida casuística no encontremos ningún caso.
Una característica muy peculiar de los tumores neonatales es el denominado "período de gracia oncológico neonatal", término que fue utilizado por primera vez por Bolande en 1985 68,69, consistente en que la mayoría de tipos histológicos tumorales neonatales presentan un mejor comportamiento biológico que el esperado en épocas pediátricas posteriores. Traduce que la edad per se actúa como un factor pronóstico importante e independiente del tipo tumoral, patrón histológico y extensión tumoral. El mecanismo subyacente implicado se denomina regresión espontánea, que se define como la desaparición parcial o completa de un tumor benigno o maligno. En 11 pacientes documentamos la regresión espontánea de la tumoración, en seis completa y en cinco parcial, no encontrando datos globales en las series publicadas.
Finalmente pensamos que los tumores neonatales debido a su rareza poblacional, deben ser abordados mediante estudios colaborativos nacionales e internacionales, unificando la metodología y los criterios diagnósticos y terapéuticos. Así podremos conocer con más detalles las características generales y específicas de las diversas variedades histológicas de los tumores neonatales.
En conclusión, los tumores neonatales, por sus características biológicas constituyen un subgrupo específico dentro de la oncohematología pediátrica. Es necesario unificar el concepto de tumor neonatal para poder analizar y comparar las diferencias de las distintas series publicadas. A pesar de las limitaciones metodológicas encontradas, las características clínicas, diagnósticas, terapéuticas y evolutivas de nuestra serie son similares a las publicadas por otros autores. Probablemente, las diferencias encontradas puedan explicarse por los diversos criterios de selección de los diferentes estudios.
Correspondencia: Dr. J. Ferrís i Tortajada.
Unidad de Salud Medioambiental Pediátrica.
Sección de Oncología Pediátrica.
Hospital Materno-Infantil Universitario La Fe.
Avda. Campanar, 21. 46009 Valencia. España.
Correo electrónico: ferris_jos@gva.es
Recibido en abril de 2005.
Aceptado para su publicación en febrero de 2006.
