



Introducción
El proceso de destete es una fase crítica en la evolución de cualquier paciente tratado con soporte ventilatorio artificial. La recuperación de la respiración espontánea se realiza paulatinamente y puede verse dificultada por numerosas causas entre las que se encuentran las enfermedades neuromusculares.
Presentamos el caso de un lactante en el que la hipotonía muscular llevó a una insuficiencia respiratoria restrictiva que hizo fracasar repetidamente la separación del paciente del respirador.
Caso clínico
Varón de 6 meses que ingresa por neumonía por Haemophilus influenzae. Tras una mejoría inicial con tratamiento antibiótico progresa hasta insuficiencia respiratoria grave hipercápnica no hipoxémica con alta demanda ventilatoria que requirió intubación. Antecedentes familiares: hermana de 9 años portadora de válvula ventrículo peritoneal por hidrocefalia congénita, colpocefalia, agenesia parcial cuerpo calloso, quiste aracnoideo e hipoacusia neurosensorial bilateral. Antecedentes personales: embarazo controlado por hidrocefalia. Cesárea a las 37 + 6 semanas. Apgar 9/10. Peso: 2.550 g (P25), PC: 37,5 cm (> P90). Ecografía trasfontanelar y resonancia magnética (RM): colpocefalia con agenesia cuerpo calloso, dilatación sistema ventricular a expensas de astas occipitales y ambos ventrículos laterales sin dilatación del III ventrículo. No requirió derivación ventriculoperitoneal de líquido cefalorraquídeo. Ingreso a los 2 meses por bronquiolitis virus respiratorio sincitial negativo.
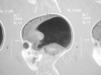
A su ingreso en la unidad de cuidados intensivos, conectado a ventilación mecánica, presenta abundante producción de esputo purulento, fiebre y leucocitosis. Radiografía de tórax: infiltrados alveolares sin condensación lobular. Tras permanecer 48 h la asistencia respiratoria, se extuba, presentando una ventilación adecuada con un índice de oxigenación de 250 y un nivel de reactividad normal. A los 2 días comienza con retención progresiva de CO2 sin estridor ni signos de trabajo respiratorio, precisando reintubación nasotraqueal. Después de 3 días se encuentra consciente y orientado con hipotonía generalizada, arreflexia, ausencia de sostén cefálico y sedestación. Respuesta patológica ante suspensión vertical y horizontal con atonía de miembros y bamboleo de cabeza, actitud "en libro abierto" con extensión pasiva de tobillos. Su situación clínica es estable con un trabajo respiratorio aparentemente fisiológico y un intercambio gaseoso normal por lo que a los 3 días se extuba por segunda vez. Aparece de nuevo hipercapnia con fracaso de respiración espontánea requiriendo reintubación. Con el diagnóstico de insuficiencia respiratoria restrictiva de origen neuromuscular secundaria a cuadro de hipotonía generalizada se realiza test del tensilón (normal) y potenciales evocados estoacústicos (hipoacusia bilateral perceptiva grave, potenciales visuales normales). Tomografía computarizada craneal y RM: colpocefalia importante (fig. 1). Cifras de creatincinasa (CK) normales. Electromiograma: patrón miopático de características no inflamatorias de distribución generalizada aunque afecta predominantemente a músculos proximales (sobre todo a extremidades inferiores) y de intensidad severa compatible con diagnóstico inespecífico de miopatía congénita.

Figura 1. RM corte sagital (agenesia cuerpo calloso).
Mientras recibe tratamiento con ventilación mecánica desarrolla una sobreinfección pulmonar de origen bacteriano y evoluciona hasta una bronconeumonía que es causa de su fallecimiento 21 días más tarde por hipoxemia refractaria.
Con la sospecha de miopatía, se realiza estudio necrópsico de biopsias de psoas, cuádriceps, musculatura intercostal y diafragma. Las dos primeras muestran sustitución adiposa sin fibras musculares. Las biopsias de musculatura intercostal presentan intensa infiltración adiposa y fascículos de fibras musculares atróficas alternando con otras normales o hipertróficas y bandas de tejido fibroso entre fascículos. El estudio ultraestructural demuestra intensa infiltración grasa, fibrosis y fibras musculares muy hipotróficas alternando con ocasionales elementos hipertróficos. Las muestras de diafragma están constituidas por fibras musculares relativamente preservadas sin signos de atrofia. El conjunto de hallazgos sugiere una atrofia muscular avanzada concordante con atrofia muscular espinal o enfermedad de Werdnig-Hoffman (AME) (fig. 2). Se realiza estudio genético de portadores de AME en los progenitores del niño mediante análisis cuantitativo mostrando ambos una dosis única del exón 7 del gen SMN1 por lo que la probabilidad de que sean portadores de la deleción del citado gen es superior al 95 % y por ello altamente probable de que el lactante fuese homozigoto para dicha mutación.

Figura 2. Biopsia muscular.
Discusión
El proceso de separación de la ventilación mecánica conocido como "destete" constituye un período crítico de límites mal definidos que comienza cuando se estima que el paciente es capaz de asumir por sí mismo, al menos parcialmente, la carga de trabajo de su ventilación alveolar. Su resultado es incierto porque durante el destete el paciente va a poner a prueba el rendimiento de los elementos que constituyen el sistema de control respiratorio: la integridad de los centros bulbares, la transmisión eficaz de los impulsos nerviosos y el rendimiento eficaz de la bomba muscular respiratoria. Todos estos elementos deben funcionar de manera coordinada para garantizar una ventilación eficaz con coste energético bajo, y el fracaso de alguno de ellos aboca inevitablemente a la insuficiencia respiratoria. Las enfermedades neuromusculares son una causa poco frecuente de insuficiencia respiratoria en lactantes. En relación con la estructura anatómica afectada pueden agruparse como enfermedades de motoneurona, enfermedades axonales y enfermedades de placa motora. La diferenciación clínica de las diferentes entidades no es sencilla en el lactante y suelen agruparse bajo la denominación de carácter descriptivo de "niño hipotónico (floppy baby)".
Entre los trastornos de la motoneurona del asta anterior de la médula capaces de provocar insuficiencia respiratoria se encuentran la AME y la poliomielitis. La fibra nerviosa resulta primariamente afectada en las leucodistrofias, intoxicaciones y polineuritis. La miastenia grave es prácticamente única causa de afectación de la unión neuromuscular en niños. En la distrofia muscular, miopatías congénitas y polimiositis el defecto asienta en la fibra muscular estriada. En nuestro paciente era evidente clínicamente la integridad funcional del centro respiratorio. La normalidad de la respuesta al tensilón y la ausencia de reflejos osteotendinosos nos permitieron descartar la miastenia grave. En esos casos la electromiografía (EMG) muestra un patrón de denervación característico que no se hallaba presente en nuestro caso. La ausencia de signos clínicos de desmielinización y la normalidad de la apariencia de la sustancia blanca en las pruebas de imagen nos permitió descartar la leucodistrofia.
La enzima más útil en el diagnóstico de las enfermedades musculares es la CK cuya concentración se eleva característicamente por la destrucción del tejido muscular que se produce en la polimiositis o en la distrofia muscular. Los niveles normales de CK, la inexistencia de atrofia muscular grave y la ausencia de un patrón EMG sugestivo nos permitieron descartar esas entidades.
Las miopatías congénitas se caracterizan clínicamente por debilidad e hipotonía desde el nacimiento y por alteraciones morfológicas características en el examen histológico y ultraestructural. En esos casos las cifras de CK son normales. La miopatía nemalínica presenta una hipotonía severa, escasa actividad espontánea y dificultad respiratoria, acompañándose de paresia facial con motilidad ocular conservada. Se asocian dismorfias como facies alargada, pectus excavatum, dolicocefalia. El diagnóstico de confirma mediante biopsia muscular, donde aparecen bastoncillos citoplasmáticos en la banda Z acumulados cerca del sarcolema. En la enfermedad de Werdnig-Hoffmann, en cambio, aparece atrofia muscular. Por tanto, solamente la biopsia muscular es capaz de establecer con seguridad el diagnóstico de la atrofia musculoespinal cuando, como ocurría en nuestro caso, los hallazgos clínicos y el EMG no son sugestivos. Las miopatías y las distrofias congénitas pueden dar las mismas manifestaciones clínicas con ausencia de reflejos osteotendinosos y con cifras normales de CK pero la afectación de la musculatura intercostal característica de la AME produce una deformidad peculiar de la caja torácica que ayuda a diferenciarla del característico aplastamiento anteroposterior que provocan las miopatías. Por otra parte la atrofia no siempre está presente en la exploración de los pacientes diagnosticados de AME.
El paciente presentaba una AME tipo I de acuerdo a la actual clasificación reuniendo los criterios de sospecha fuera del período neonatal de Rudnik-Schoneborn, exceptuando la existencia de fasciculaciones linguales.
De acuerdo a Hendricks et al, creemos que el caso también se ajusta a la descripción realizada del síndrome de Chudley-McCullough OMIN 604213, sin que en la actualidad se conozca el locus cromosómico implicado aunque sí el patrón de herencia mendeliana autosómico recesiva que reúne la familia estudiada al presentar una hermana con las mismas alteraciones encefálicas y unos padres sanos que pertenecen a una localidad en la que hasta hace 3 décadas existía alta tasa de endogamia.
Por tanto, nuestro caso ilustra la gran dificultad diagnóstica de algunas enfermedades neuromusculares, en las que sólo un alto índice de sospecha y la exclusión de otros procesos llevan al clínico a solicitar una biopsia muscular y estudio genético.
Correspondencia: Dra. L. Ruiz Pérez.
Hospital General Universitario de Alicante. Servicio de Pediatría.
Maestro Alonso, 109. 03010 Alicante. España.
Correo electrónico: lorearuiz@hotmail.com
Recibido en octubre de 2005. Aceptado para su publicación en julio de 2006.
