




Oviedo, 5-6 de mayo de 2005
Pósters
TIROIDES
Dos casos de bocio neonatal hipotiroideo asociados a patología tiroidea materna
J. Guerrero Fernández, E. Martín Campagne, B. Lodoso Torrecilla, M. Álvarez Acevedo, M.M. Martínez López y R. Gracia Bouthelier
Servicio de Endocrinología Infantil. Hospital Infantil La Paz. Madrid. España.
Introducción: La presencia de un bocio hipotiroideo en el período neonatal responde a muy pocas causas, y la mayoría son secundarias a patología tiroidea materna. Como ejemplo representativo de esta afirmación, presentamos dos recién nacidos con bocio, hijos de madres con patología tiroidea intragestacional.
Casuística:Caso 1: Neonato, nacido a la semana 29 de edad gestacional (peso y longitud acordes), que presenta bocio visible y palpable no compresivo. Antecedentes obstétricos: madre en tratamiento sustitutivo con levotiroxina por hipotiroidismo autoinmune desde hace 3 años; a la semana 26 de gestación se detecta polihidramnios secundario a bocio fetal compresivo, por lo que se trata con levotiroxina vía amniótica. Período neonatal: se detecta, a nivel umbilical, TSH de 25 U/ml y T4 libre de 0,99 μg/dl con anticuerpos antitiroglobulina de 815 UI/ml (VN: < 60) y antiperoxidasa de 96,18 UI/ml (VN: < 60); ecografía tiroidea: ambos lóbulos aumentados de tamaño y homogéneos sin aparente compresión sobre traquea ni esófago; es tratado con levotiroxina a 8 μg/kg/día. Controles posteriores: a los 8 días de vida: TSH: 0,39, T4 libre: 1,8; a los 13 días de vida: normalización clínica y ecográfica del tamaño tiroideo; a los 39 días precisa nuevo aumento de levotiroxina ante una TSH de 39; finalmente, con tres meses de edad, se suspende tratamiento sustitutivo tras detectarse niveles suprimidos de TSH. En la actualidad, con cuatro meses de vida y en ausencia de tratamiento sustitutivo, presenta normalidad tiroidea clínica y analítica, incluidos los anticuerpos antitiroideos. Caso 2: Neonato, nacido a término con peso y longitud adecuados para su edad gestacional, que presenta bocio visible y palpable pero no compresivo. Antecedentes obstétricos: hipertiroidismo materno no autoinmune (antecedente de bocio multinodular) detectado a la semana 15 de edad gestacional. Se inicia tratamiento con propiltiouracilo a 25 μg. Ecografías sin alteraciones en el feto. Período neonatal: se detecta, al nacimiento, TSH > 200 U/ml y T4 libre de 0,78 μg/dl con anticuerpos antitiroideos y TSI negativos, por lo que se instaura tratamiento sustitutivo con levotiroxina a 8 μg/kg/día. A los dos meses, ante un descenso rápido de la TSH hasta concentraciones indetectables, se suspende definitivamente el tratamiento. En la actualidad, con tres meses de edad, presenta normalización clínica y analítica del sistema hipófiso-tiroideo.
Discusión: La etiología del bocio neonatal hipotiroideo incluye diversas causas: inmadurez de la glándula tiroidea (pretérminos), el inducido por la ingesta materna de fármacos antitiroideos, el secundario a dishormonogénesis, el desencadenado por autoanticuerpos maternos (antitiroideos o antirreceptor de TSH bloqueantes) y, finalmente, el debido a la ingesta materna excesiva de yodo por efecto Wolf-Chaikoff o a su déficit. El primer caso responde a un origen inmunológico que representa el 2 % de los hipotiroidismos congénitos. Con respecto a su patogenia no hay consenso sobre si concentraciones elevadas de anticuerpos antitiroideos maternos suponen a un mayor riesgo de padecer hipotiroidismo congénito, o bien, si este riesgo viene dado por el grado de actividad de dichos anticuerpos más que por su número. El tratamiento sustitutivo resulta necesario hasta que los anticuerpos antitiroideos de origen materno desaparezcan. El segundo caso obedece al efecto antitiroideo del propiltiouracilo que, a diferencia de la T3, T4 libre y TSH maternas, atraviesa fácilmente la placenta. Probablemente la facilidad con que se gestó un hipotiroidismo fetal se debía a que la madre no presentaba enfermedad de Graves y, por tanto, no había factores, como la estimulación inmunológica de los receptores tiroideos fetales, que compensaran el efecto inhibitorio del antitiroideo. El tratamiento sustitutivo, a diferencia de lo que se discute en el tratamiento del hijo de madre con Graves-Basedow, era igualmente necesario mientras permaneciera el antitiroideo en la sangre del recién nacido.
Tiroiditis autoinmune en la infancia. Revisión casuística
A. Vela Desojo, C. Poza, A. Aguayo, I. Rica, M.A. Aniel Quiroga y P. Martul
Servicio de Pediatría. Endocrinología Infantil. Hospital de Cruces. Barakaldo. Bilbao. España.
Introducción: La tiroiditis de Hashimoto es una enfermedad autoinmune frecuente en la infancia. Además de la predisposición genética existen otros factores ambientales que favorecen la aparición de la enfermedad.
Objetivo: Estudiar los pacientes con tiroiditis controlados en la consulta de Endocrinología infantil. Analizar los antecedentes, factores predisponentes, inicevolución de la enfermedad.
Pacientes y métodos: Estudio retrospectivo de los niños controlados por tiroiditis en los 8 últimos años en la consulta de Endocrinología infantil.
Resultados: Se han estudiado 47 niños (41 niñas y 6 niños). La edad media al diagnóstico es de 10 años (r: 5,5-15,1). Con un Índice de Masa Corporal (SDS): ζ:0,46 (r:1,43-6,04) sólo dos pacientes tenían un IMC > 2 SDS. La Talla (sds): ζ: 0,27 (r: 2,3-6,4). Ningún paciente tomaba medicaciones de forma crónica. Cuatro pacientes presentaron un peso y/o talla al nacimiento < P10. En los que se conocía el dato el 50 % de los padres eran fumadores. El 20 % de los pacientes tenía antecedentes familiares de tiroiditis. El motivo de consulta fue: 66 % bocio, 20 % retraso de crecimiento, 8 % dolor de cuello y 6 % otros (vitíligo, angioedema, pubertad precoz, obesidad). Desde el punto de vista de la función tiroidea el 47 % tenían hipotiroidismo, 17 % hipotiroidismo subclínico, 34 % eutiroidismo y 2 % hipertiroidismo. Los niveles de TSH (μU/ml) en los casos de hipotiroidismos fueron: ζ: 115 (r: 5-837). La media de l-tiroxina al diagnóstico fue de 1,3 μg/kg/día que es la misma con la que se consigue estabilidad. La evolución de la enfermedad se mantiene estable en el 75 %; el 21 % empeoran y un paciente mejora. Ningún paciente ha presentado complicaciones posteriores al diagnóstico.
Conclusiones:1) No encontramos más incidencia de crecimiento intrauterino retardado que en la población general. 2) La mayoría de los pacientes son mujeres. 3) No se constata una mayor exposición a drogas o tabaco como desencadenante de la enfermedad. 4) La curación es excepcional. 5) La obesidad no es un signo típico de la tiroiditis de Hashimoto.
Valoración de la efectividad de un programa de diagnóstico precoz y seguimiento del hipotiroidismo congénito: crecimiento y maduración somáticos (I)
A. Gibert Agulló, E. Vicens-Calvet, N. Potau Vilalta, M.ª Albisu Aparicio y A. Carrascosa
Servicio de Endocrinología. Hospital Materno-Infantil Vall d'Hebron. Barcelona. España.
Objetivo: Estudiar el crecimiento (talla y peso) y maduración somática (edad ósea e inicio de la pubertad) en 136 casos (106 niñas, 30 niños) de hipotiroidismo congénito controlados en la unidad de seguimiento del hipotiroidismo congénito del hospital X (1986-1997).
Material y métodos: estudio longitudinal y retrospectivo de los casos de hipotiroidismo congénito nacidos durante el período 1986-1997, diagnosticados y tratados en la unidad de seguimiento del hipotiroidismo congénito del hospital X. Crecimiento: cada 6 meses se han valorado talla, SDS talla, VC, SDS VC, peso, IMC y SDS IMC. Para cada período de 6 meses se han considerado los valores registrados entre 3 meses antes y 3 meses después de la edad cronológica (EC) central del período. Maduración ósea: se han evaluado todas las radiografías practicadas centrándolas según el mismo criterio. Inicio de la pubertad: se ha valorado la edad de aparición del estadio B2 en niñas y de un volumen testicular ≥ 4 ml en los niños. La edad se ha centrado en el período máximo de 6 meses con relación a la visita anterior. Se ha analizado también la edad en la que tiene lugar el take off o despegue, obtenida a partir de la representación gráfica de la VC. Estándares de referencia: Talla: Tanner-Whitehouse 1966, Carrascosa 2004. Peso e IMC: Roland-Cachera 1982, Carrascosa 2004. Edad ósea: G&P. Pubertad: Marshall-Tanner 1969-70.
Resultados:Talla: se sitúa hasta el inicio de la pubertad (niños y niñas) + 0,5 SD respecto a Tanner. Esta diferencia es estadísticamente significativa en la mayoría de los puntos, especialmente en las niñas. Respecto a los estándares de Carrascosa no hay diferencias significativas (niños y niñas). Velocidad de crecimiento: respecto a los estándares de Tanner, en las niñas no existen diferencias significativas hasta los 9 años y posteriormente se sitúan por encima y luego por debajo de dichos estándares. En los niños no se observan diferencias respecto a Tanner. Peso e IMC: el IMC, tanto para los niños como para las niñas, se sitúa + 1 SD respecto a Roland-Cachera con diferencias estadísticamente significativas. El peso respecto a Carrascosa no muestra diferencias estadísticamente significativas. Maduración ósea: sigue una evolución paralela a la EC. Inicio de la pubertad: en el caso de las niñas se observa un adelanto de la edad del despegue de la VC de 1,17 años respecto a los estándares de Marshall y Tanner. Entre los 9 y 11 años hay mayores porcentajes de niñas con HC que presentan B2 que en el grupo de Marshall y Tanner. Estas diferencias son estadísticamente significativas e indican que las niñas de este estudio inician la pubertad antes que las de Marshall y Tanner. Estos datos pueden explicarnos las diferencias observadas en la VC. Al ser menor el número de niños no pueden realizarse valoraciones estadísticas.
Conclusiones: los niños y niñas del presente estudio siguen un crecimiento y maduración ósea igual al de la población actual (Carrascosa, 2004) y superior a los estándares de Tanner (1966) y Roland-Cachera (1982). El inicio de la pubertad en las niñas tiene lugar antes que el referido por Marshall y Tanner (1969-1970). Globalmente, estos resultados indican normalidad en el desarrollo respecto a la actual población de referencia.
Valoración de la efectividad de un programa de diagnóstico precoz y seguimiento del hipotiroidismo congénito: desarrollo psicológico (II)
A. Gibert Agullo, E. Vicens-Calvet, M. Bargada Esteve, N. Potau Vilalta, M.ª Albisu Aparicio y A. Carrascosa Lezcano
Servicio de Endocrinología y Paidopsiquiatría. Hospital Materno-Infantil Vall d'Hebron. Barcelona. España.
Objetivo: estudiar la evolución psicológica de 51 casos de hipotiroidismo congénito controlados en la unidad de seguimiento del hipotiroidismo congénito del hospital X (1986-1997) y su relación con la gravedad y duración del hipotiroidismo neonatal.
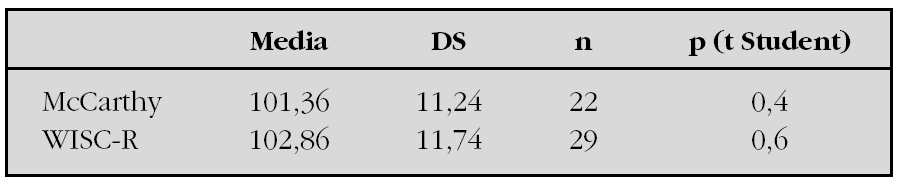
Material y métodos: estudio transversal y retrospectivo de 51 casos de hipotiroidismo congénito nacidos durante el período 1986-1997, diagnosticados y tratados en la unidad de seguimiento del hipotiroidismo congénito del hospital X. 1) Gravedad y duración del hipotiroidismo congénito: T4 y TSH en la primera visita (inicio del tratamiento). Índice clínico de Dussault. Superficie del núcleo de osificación inferior del fémur. Días de vida en la primera visita y al alcanzar el T4 el valor 10 μg/dl. 2) Tests psicológicos: McCarthy: a los 4-5 años. WISC-R: a partir de los 6 años. Población de referencia: McCarthy y WISC adaptado a la población española (TEA Ediciones, Madrid 1988).
Resultados:1) Los resultados de los tests de WISC-R y McCarthy se detallan en la siguiente tabla.
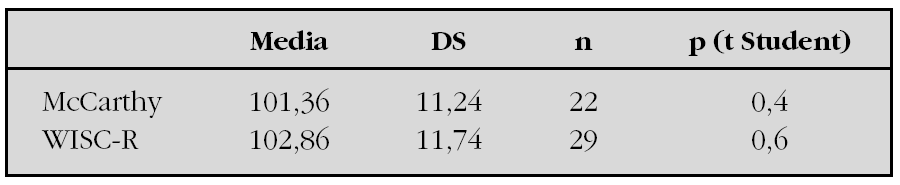
2) Correlaciones: WISC-R: correlación negativa con los días de vida en alcanzar el T4 el valor 10 μg/dl (R = 0,46, p = 0,01). McCarthy: correlación positiva con los niveles de T4 al inicio del tratamiento (R = 0,47, p = 0,04). 3) La tendencia en el resto de correlaciones, sin llegar a ser estadísticamente significativas, es en general, una menor puntuación en los tests psicológicos cuanto mayor es la gravedad y duración del hipotiroidismo neonatal.
Conclusiones: Los niños y niñas del presente estudio tienen, como grupo, un cociente intelectual que no difiere del de la población de referencia. Se observa que la puntuación en los tests psicológicos es menor cuanto mayor fue la gravedad y duración del hipotiroidismo neonatal.
Evaluación de la función tiroidea, anticuerpos antitiroideos y yoduria durante el embarazo
A. Aguayo, G. Grau, M. Espada, M.A. Aniel-Quiroga, I. Ocerin, A. Vela, I. Rica, J.R. Bilbao, J.I. Pijoan, J.C. Vitoria, A. Martin-Pagola, L. Castaño y P. Martul
Servicio de Endocrinología Pediátrica. Laboratorio Hormonas. Unidad Investigación. Ginecología. Epidemiología Clínica. Gastroenterología Infantil. Hospital de Cruces y Laboratorio Normativo de Salud Pública. Bilbao. España.
Introducción: Estudios previos han mostrado que concentraciones bajas de tiroxinemia o yoduria en mujeres durante el embarazo se correlacionan directamente con una disminución del cociente intelectual de sus hijos. El objetivo es evaluar de forma prospectiva la función tiroidea y yoduria durante el embarazo en mujeres que previsiblemente tendrán el parto en el hospital terciario donde se realiza el estudio.
Material y métodos: Se contactó con los ginecólogos y matronas de los centros de salud del área del citado hospital para obtener su colaboración. Tras obtener su consentimiento informado se recogió una muestra de sangre y otra de orina al final del 1.er y 2.º trimestre del embarazo (coincidiendo con las extracciones de la rutina habitual). Se presentan los estudios realizados durante los primeros 32 meses. Se determinaron los niveles de tiroxina libre (T4), TSH y anticuerpos anti-TPO en plasma por enzimoinmuno-ensayo por quimioluminiscencia (Immulite 2000). La yoduria se determinó por cromatografía líquida de alta resolución (HPLC).
Resultados: Se presentan los resultados de 3603 muestra s sanguíneas y 3.560 de orina. Para evitar distorsiones con relación a la diuresis los resultados de yoduria se expresan como μg de yodo por gramo de creatinina. En el primer trimestre las yodurias por debajo de los límites recomendados (200 μg) llegan al 90 % y las muy deficitarias (< 100 μg) alcanzan el 59 %. Siendo la mediana de 89 μg en el 2.º trimestre los valores son 68 y 25 %, respectivamente, y la mediana de 154 μg. En cuanto a los niveles de T4 libre la media del 1.er trimestre fue 1,34 ng/dl. En el 2.º trimestre fue de 1,1 ng/dl. Respecto a la TSH en el 1.º trimestre z = 1,79 mU/l y en el 2.º trimestre = 2,04 mU/l. Las diferencias entre el 1.º y 2.º trimestre son estadisticamente significativas en todos los parametros estudiados. Finalmente los anticuerpos anti-TPO están elevados en un 10 % de las mujeres el 1.er trimestre y un 7,9 % el 2.º trimestre. Un 7,0 % de las mujeres los tenían elevados en ambos trimestres. Un total de 18 mujeres tenían una cifra por debajo del percentil 10 de T4 en ambos trimestres y 11 tenían TSH por encima de 5,0 mU/l.
Conclusiones:1) Existe una severa deficiencia de yodo en las embarazadas estudiadas y aunque mejora en el 2.º trimestre del embarazo la situación sigue siendo inaceptable. 2) Las cifras medias de T4 libre y TSH están dentro de los límites de la normalidad con un descenso de T4 y elevación de TSH en el 2.º trimestre. 3) En el 2.º trimestre disminuye el número de mujeres embarazadas con anticuerpos anti-TPO. 4) Es preciso realizar una activa y agresiva campaña de yodación en la zona donde se ha realizado este estudio con particular énfasis en los períodos de preembarazo y primeros meses de la gestación.
Este trabajo se ha realizado con ayudas del Departamento de Sanidad del Gobierno Vasco, de la Fundación Salud 2000, de la Red de Centros del FIS (RCMN C03-08) y de la Fundación Ikertu.
Enfermedad de Graves neonatal
J.M. Martos Tello, A. Escribano Muñoz y A. Gutiérrez Macías
Servicio de Pediatría. Hospital Universitario Virgen de La Arrixaca. Murcia. España.
Introducción: La tirotoxicosis neonatal es una entidad rara que representa menos del 1 % de todos los casos de hipertiroidismo en la infancia. La frecuencia de esta enfermedad en los hijos de mujeres hipertiroideas embarazadas varía el 0,5-9 %. Normalmente se manifiesta de forma transitoria, aunque se acepta una forma permanente de la misma. Contribuir en el manejo y tratamiento de estos neonatos a través de este caso constituye nuestro objetivo.
Caso clínico:Motivo de consulta: Neonato de 15 días con sospecha de hipertiroidismo. Antecedentes familiares y personales: Madre de 36 años. Prima hermana materna hipotiroidea. Gestación controlada. Diagnóstico de enfermedad de Graves en el 3er mes por taquicardia materna. En tratamiento con propiltiouracilo 50 μg cada 8 h y diazepam con buen control. En ecografías prenatales no se detectó taquicardia fetal. Parto eutócico, Apgar 9/10. PN: 3.400 g. Período neonatal inmediato normal. Enfermedad actual: Acuden a consulta de endocrinología infantil por nerviosismo, irritabilidad, sueño lábil y gran avidez por el alimento. Aportan resultados de analíticas: 6.º día de vida: TSH > 100 μUI/ml (0,3-4,5); T4L: 0,64 ng/dl (0,9-1,7); T3L: 9,7 pg/ml (2,5-4,4); 14.º día de vida: TSH 0,084 μUI/ml (↓ ↓ ↓); T4L 7,49 ng/d l (↑); T3L: 25,2 pg/ml (↑ ↑). Exploración física: P: 3,380 g. BEG, buena coloración e hidratación, piel algo sudorosa. Cráneo normoconfigurado, FA 2 x 3 NT, exoftalmos leve, retracción palpebral discreta, bocio grado II, telarquia fisiológica neonatal. ACP: taquicardia (180 lpm), no soplos ni estertores, resto de exploración física normal. Evolución y tratamiento: Con el diagnóstico de tirotoxicosis neonatal iniciamos tratamiento con solución de lugol 1 gota/8 h; metimazol 0,5 mg/kg/día y propanolol 1 mg/kg/día. Se realizan controles clínicos y analíticos semanal o quincenalmente, monitorizando tanto hormonas tiroideas como TSI (que resultan positivos en la primera determinación), así como hemogramas seriados para descartar efectos secundarios de la medicación. Precisa asociación de tiroxina con 15 días de tratamiento por presentar hipotiroidismo bioquímico. Progresiva negativización de TSI. Suspensión de tratamiento con 9 semanas de vida, coincidiendo con normalización tanto analítica como clínica. No se aprecia en ningún momento efectos secundarios por el tratamiento.
Discusión: Como puede apreciarse por los resultados analíticos, tras el nacimiento se produce un estado de hipotiroidismo transitorio debido probablemente a varios factores: paso de anticuerpos bloquedores maternos (TBI); paso de anticuerpos antitiroglobulina y microsomal y efecto de antitiroideos administrados a la madre. Pocos días después desarrolla hipertiroidismo debido al paso de TSI maternos. Usando los distintos índices pronóstico para el desarrollo de hipertiroidismo en el neonato podría haberse previsto esta situación.
Conclusiones: El hipertiroidismo neonatal es una entidad rara secundaria en la mayoría de los casos al paso de anticuerpos TSI maternos. Puede asociarse a hipotiroidismo transitorio. Mediante la determinación de índices de predicción es posible determinar la posibilidad de sufrir hipertiroidismo en neonatos hijos de madres con Graves. El tratamiento de la enfermedad de Graves neonatal debe ser precoz, realizarse con lugol y anititiroideos de síntesis y mantenerse hasta la negativización de los niveles de TSI.
Enfermedad tiroidea autoinmune y gastritis autoinmune
D. Crespo Marcos 1, P. Chimenti Camacho 1, B. Riaño Méndez 1, M.I. Marsinyach Ros 1, N. López Lazareno 2, M. Rodríguez Mahou 3, A. Rodríguez Sánchez 1 y M.D. Rodríguez Arnao 1
1Unidad de Metabolismo y Desarrollo. Servicios de 2Bioquímica e 3Inmunología. Hospital General Universitario Gregorio Marañón. Madrid. España.
Introducción: Se ha descrito una elevada prevalencia (de hasta el 30 %) de anticuerpos anticélula parietal gástrica (PCA) en niños con enfermedad tiroidea autoinmune. La positividad de los PCA junto con la elevación de los niveles de gastrina plasmática constituyen un fiable indicador de gastritis autoinmune. El objetivo de nuestro trabajo fue determinar la prevalencia de estos marcadores en niños y adolescentes diagnosticados de enfermedad tiroidea autoinmune.
Sujetos y métodos: Incluimos 26 pacientes (5 varones) con enfermedad tiroidea autoinmune (3 de ellos con enfermedad de Graves). Fueron diagnosticados en rango de hipofunción, hiperfunción o normofunción tiroidea por presentar anticuerpos antiperoxidasa tiroidea (anti-TPO) y/o antitiroglobulina (anti-Tg) muy elevados. En los hipertiroideos, se determinó también la presencia de anticuerpos estimulantes del receptor de TSH (TSI), con resultados positivos. Se extrajeron muestras basales de los pacientes para realizar: hemograma (Coulter); concentraciones plasmáticas de vitamina B12 y ácido fólico (quimioluminiscencia), gastrina (radioinmunoanálisis); y determinación de PCA (inmunofluorescencia indirecta). Así mismo, se estudiaron la edad en el momento de las determinaciones y la comorbilidad de otras patologías autoinmunes.
Resultados: La edad cronológica (media ± DS) de los pacientes en el momento de la extracción fue de 14,21 ± 3,18 años; rango: 6,33-19,58 años). Los niveles de T4 libre (N: 0,9-2,0 ng/dl) y TSH (N: 0,5-4,5 μU/ml) se encontraban en niveles normales con tratamiento hormonal sustitutivo con L-tiroxina oral (en los tres pacientes con enfermedad de Graves, asociada a metimazol). No se detectaron alteraciones en la hemoglobina de ninguno de los 26 pacientes, y además se encontraron los valores de volumen corpuscular medio (VCM) y hemoglobina corpuscular media (HCM) dentro de límites normales. Había seis casos de diabetes mellitus y dos de enfermedad celíaca. Hallamos un paciente con PCA positivos, en una niña hipertiroidea de 14 años, en la que no se asociaban otras patologías autoinmunes ni alteraciones en ninguno de los demás parámetros estudiados. Las concentraciones plasmáticas obtenidas (media ± DS) fueron: vitamina B12 (461,36 ± 226,98 pg/ml; N: 200-980 pg/ml), ácido fólico (9,2 ± 4,37 ng/ml; N: 2,5-15 ng/ml) y gastrina (68,22 ± 34,35 ng/ml; N: 18-185 ng/ml).
Conclusiones:1) La enfermedad tiroidea autoinmune de comienzo en infancia y adolescencia se asocia a otras patologías de similar etiología, siendo las de mayor prevalencia diabetes mellitus y enfermedad celíaca. 2) La gastritis autoinmune puede estar presente en estos pacientes de forma asintomática, no pudiendo ser excluida basándose en la normalidad de los valores de hemoglobina, VCM y HCM. 3) La determinación de PCA constituye un marcador precoz y sensible para la detección de esta enfermedad. 4) En los pacientes diagnosticados de enfermedad tiroidea autoinmune deben estudiarse periódicamente los niveles de vitamina B12, ácido fólico y gastrina, así como determinar la presencia de PCA, como marcadores de gastritis autoinmune asociada. 5) La gastritis autoinmune se detecta con mayor frecuencia en pacientes con enfermedad tiroidea autoinmune hiperfuncionante, pudiendo además presentarse sin comorbilidad con otras patologías autoinmunes. 6) En los casos en los que se detectan PCA e hipergastrinemia está indicada la realización de biopsia gástrica para confirmación diagnóstica.
Función tiroidea neonatal en hijos de madres con patología tiroidea
M.T. Jiménez Fernández, E. Mayayo Dehesa, J.I. Labarta Aizpun, G Terrer Manrique y A. Ferrández Longás
Servicio de Pediatría. Unidad de Endocrinología. Hospital Universitario Miguel Servet. Zaragoza. España.
Introducción: La enfermedad tiroidea autoinmune materna puede causar hipotiroidismo o hipertiroidismo congénito en sus hijos por paso transplacentario de anticuerpos antitiroideos. En muchas ocasiones las madres desconocen la causa de su trastorno. Ello ha motivado que se hayan controlado a todos los recién nacidos en el hospital cuyas madres padezcan o hayan padecido cualquier tipo de patología tiroidea. Se presenta la casuística correspondiente al período: enero 1988-enero 2004.
Material y métodos: 257 recién nacidos (144 varones; 113 mujeres) de 257 madres, de las cuales 127 (49,1 %) padecieron enfermedad tiroidea autoinmune (74 tiroiditis y 56 enfermedad de Graves); 96 (37,1 %) hipotiroidismo de causa no filiada; 23 (8,9 %) nódulos; 5 (1,9 %) carcinoma; 4 (1,5 %) bocio normofuncional; 1 (0,3 %) adenoma y 1 (0,3 %) bocio coloide. A los niños se les determinó las concentraciones séricas de FT4, TSH y anticuerpos antitiroideos (TPO, TG, TSI) a las 48 h; 7-10 días y 4-6 semanas de vida. La función tiroidea se catalogó en seis grupos según los valores de la población de referencia (± 2 DE para FT4 y rango de 0,3-5,5 μU/ml para TSH): eutiroidismo, hipertirotropinemia, hipotiroidismo, hipotiroxinemia, hipertiroidismo e hipertiroxinemia. En ningún parto se utilizó povidona yodada.
Resultados y conclusiones:1) En los hijos de madres con enfermedad tiroidea autoinmune: se ha constatado alta prevalencia de disfunción tiroidea (41,7 %) tanto en madres con tiroiditis (43,2 %) como con enfermedad de Graves (39,6 %). La más prevalente ha sido la hipertirotropinemia transitoria (29,1 %), de las cuales, en el 59 % la TSH fue superior a 10 μU/ml, seguida de hipotiroxinemia (5,5 %). Se han detectado 5 casos de hipotiroidismo (3,9 %), siendo uno de ellos permanente, que han sido leves o compensados y no se detectaron en el screening neonatal. La prevalencia del hipertiroidismo ha sido del 1,57 % y no ha producido manifestaciones clínicas ni ha precisado tratamiento. El título de anticuerpos antitiroideos ha sido positivo en el 52,5 % de los casos de disfunción tiroidea. 2) En los hijos de madres con hipotiroidismo de causa no filiada y en los hijos de madres con otras patologías tiroideas se ha observado, respectivamente: prevalencia de disfunción tiroidea del 31,2 y del 40 % (hipertirotropinemia transitoria: 28,1 y 36,6 %) y positividad de anticuerpos antitiroideos en el 16,7 y 11,7 %. No se ha detectado ningún caso de hipotiroidismo ni de hipertiroidismo. 3) En la disfunc. No obstante, queda por aclarar si en unos casos y otros subyace un estado de deficiencia de yodo.
Hemangiomas masivos como causa de hipotiroidismo primario
P. Chimenti Camacho, D. Crespo Marcos, P. Miliante, B. Arias Nova, B. Riaño Méndez, A. Rodríguez Sánchez y M.D. Rodríguez Arnao
Unidad de Metabolismo y Desarrollo. Hospital General Universitario Gregorio Marañón. Madrid. España.
Introducción: Los hemangiomas son tumores benignos frecuentes en la infancia; se caracterizan por un período de rápido crecimiento posnatal, seguido por una fase de estabilización y posterior involución espontánea. Pueden ser asintomáticos o ser de gran tamaño, actuando como una fístula arteriovenosa que causa insuficiencia cardíaca de alto gasto, plaquetopenia y coagulopatía de consumo por agregación de plaquetas y formación de microtrombos en los vasos anómalos. Los hemangiomas masivos pueden asociar hipotiroidismo transitorio severo por aumento de actividad de la 3-yodotironina-desyodasa en el tejido del hemangioma, necesitando dosis altas de levotiroxina para normalización de la función tiroidea, con regresión espontánea del hipotiroidismo con la involución del hemangioma.
Caso clínico 1: Recién nacida a término de elevado peso para la edad gestacional (40 semanas/4.820 g). Presenta angioma plano en labio superior y filtrum, exantema petequial en tronco y miembros superiores, hepatomegalia de 3 cm, masa en hipocondrio izquierdo de 5 cm de diámetro y masa subcutánea blanda en región lumbar izquierda de 7 cm de diámetro. Asintomática. Diagnosticada de hemangioma gigante toracoabdominal y subcutáneo mediante ecografía, TC y RM. Precisó tratamiento con interferón y transfusión de concentrado de hematíes y plaquetas. Prueba de detección precoz de hipotiroidismo congénito normal (TSH 15,0 μU/ml y T4 total 9,6 μg/dl a las 48 h de vida). A los 7 días de vida presenta TSH 32 μU/ml y T4 total 6,4 μg/dl. A los 11 días de vida se confirma hipotiroidismo primario con T4L 0,4 ng/dl y TSH > 100 μU/ml y se inicia tratamiento con levotiroxina a 10 μg/ kg/día con posterior normalización de la función tiroidea.
Caso clínico 2: Recién nacido pretérmino de adecuado peso para la edad gestacional (36 semanas/2.850 g) que ingresa en neonatología por estrés respiratorio inmediato. Presenta angiomas cutaneos y hepatomegalia. Diagnosticado de angioma hepatocutáneo mediante ecografía y RM. Precisa tratamiento con inotrópicos, diuréticos y ventilación mecánica por insuficiencia cardíaca, prednisona a 5 mg/kg/día y transfusión de hematíes. Prueba de detección precoz de hipotiroidismo congénito normal (TSH 3,0 μU/ml a las 48 h de vida). Controles periódicos de función tiroidea; a los 21 días de vida presenta T4L 1,5 ng/dl con TSH 9,6 μU/ml iniciándose tratamiento con levotiroxina a 15 μg/kg/día con posterior normalización de la función tiroidea.
Comentarios: Los hemangiomas masivos son tumores vasculares raros en la infancia que asocian alta morbimortalidad. Son causa de hipotiroidismo primario adquirido transitorio por aumento de actividad de la 3-yodotironina-desyodasa en el tejido del hemangioma. El diagnóstico de confirmación etiológica se realizaría mediante biopsia del tumor para comprobar el
aumento de actividad de la 3-yodotironina-desyodasa en el tejido del hemangioma, pero no esta justificada su realización por el alto riesgo asociado. Al ser un hipotiroidismo adquirido las pruebas de detección precoz suelen ser normales, y son necesarias determinaciones periódicas de la función tiroidea durante la fase de crecimiento tumoral. A pesar de tratarse de un hipotiroidismo transitorio, es necesario un diagnóstico precoz y un tratamiento agresivo para evitar el daño neurológico. Los pacientes presentados precisaron dosis de 10 a 15 μg/kg/día de levotiroxina para normalización de la función tiroidea, valores inferiores a los referidos en la literatura (25 μg/kg/día). Si el paciente supera la fase de proliferación inicial, la evolución natural es la involución espontánea del hemangioma con normalización de la función tiroidea.
Hemiagenesia tiroidea
M. Calles Ledesma, J. Alcón, R. Tomasini, G. Dodino, D. Yeste, G. Enríquez, M. Gusssinyé y A. Carrascosa
Endocrinología Pediátrica. Hospital Materno Infantil Vall d'Hebron. Barcelona. España.
Introducción: La hemiagenesia tiroidea es una rara malformación congénita caracterizada por la ausencia o hipoplasia de uno de los lóbulos tiroideos. Es más frecuente su localización en el lado izquierdo. El diagnóstico suele realizarse de forma casual, mediante ecografía tiroidea, en el estudio de concentraciones plasmáticas elevadas de TSH o por la existencia de un bocio unilateral. Es frecuente la hipertrofia compensadora del único lóbulo existente. La función tiroidea suele estar conservada en la mayoría de los casos, aunque un porcentaje elevado de pacientes presenta cifras de TSH superiores a sujetos con tiroides íntegros.
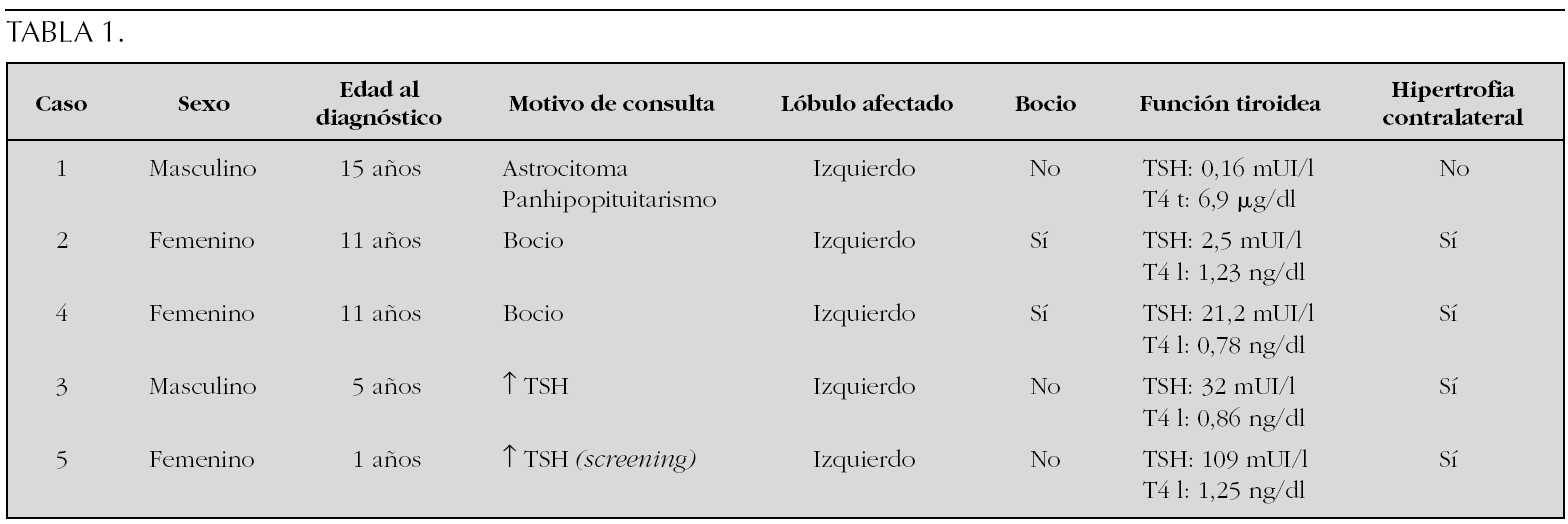
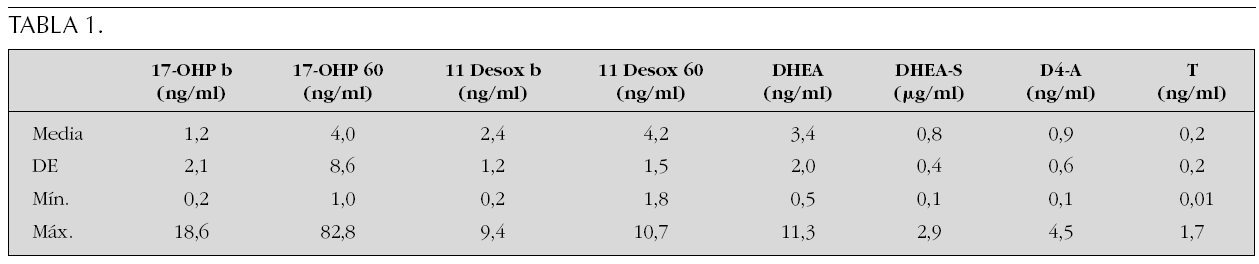
Sujetos y Método: Se revisan las características clínicas y hormonales de cinco pacientes en edad pediátrica diagnosticados en nuestro centro en los últimos 15 años. En la tabla 1 se muestran los datos clínicos, resultados de la ecografía tiroidea y de la función tiroidea de los pacientes.
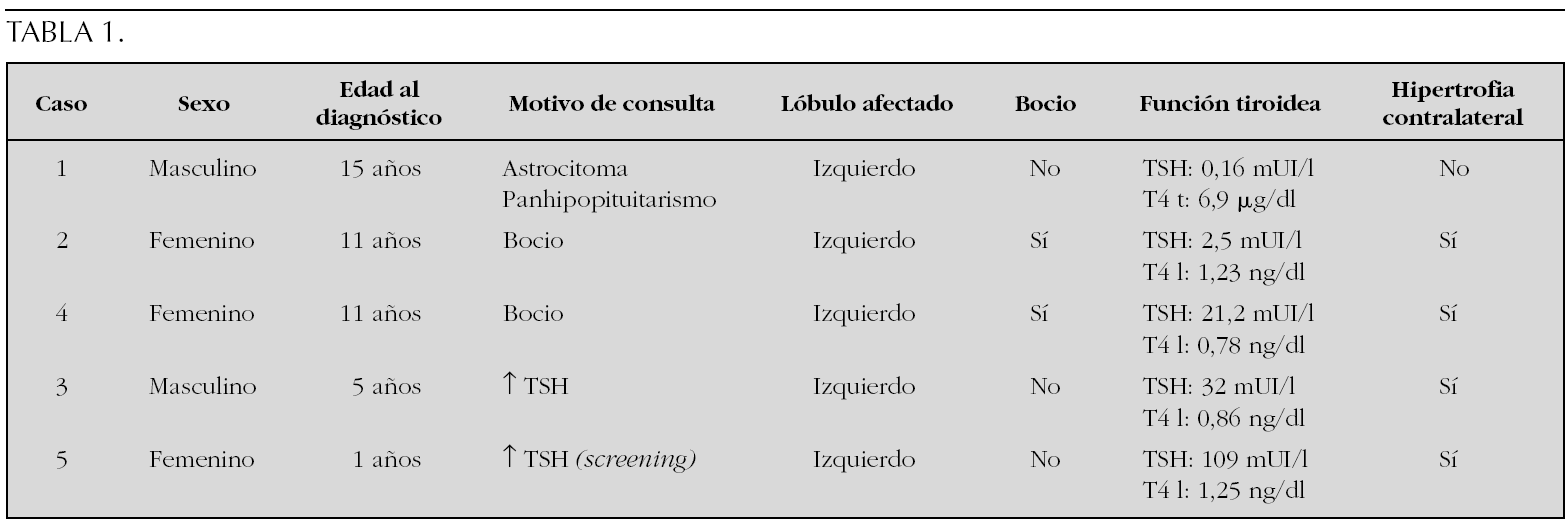
Conclusiones: El diagnóstico de la hemiagenesia tiroidea se suele realizar de forma casual, al indicarse la práctica de una ecografía de tiroides en el estudio de un paciente con concentraciones plasmáticas elevadas de TSH, o bien al detectarse la presencia de un bocio unilateral en el examen físico. El diagnóstico de esta entidad obliga a mantener un estrecho control de la función tiroidea a largo plazo.
Hipertiroidismo en la edad pediátrica
J.M. Rius Peris, M.ª F. Moreno, V. Albiach, C. Carles e I. Tarazona
Endocrinología Pediátrica. Hospital Infantil La Fe. Valencia. España.
Introducción: La causa más frecuente de hipertiroidismo en la infancia es la enfermedad de Graves (EG), en el contexto de la enfermedad tiroidea autoinmune. Se caracteriza por la hiperfunción tiroidea, el bocio y, con menor frecuencia que en el adulto, por el exoftalmos.
Pacientes y método: Estudio retrospectivo de revisión de casos, entre enero de 1987 y febrero de 2005. Se analizan: sexo, edad al diagnóstico, patología asociada, antecedentes familiares, clínica, estudio analítico, pruebas de imagen y tratamiento.
Resultados: Presentamos 20 pacientes diagnosticados de hipertiroidismo. De ellos, 2 corresponden a casos de hipertiroidismo neonatal transitorio que requirieron tratamiento. De los 18 restantes, 1 parece corresponder a un hipertiroidismo no inmune y el resto a EG. El predominio femenino es de 5/1 y la edad media al diagnóstico de 8,7 años (rango: 2,1-14,6). De los 15 pacientes en los que constan antecedentes familiares, 9 presentan antecedentes de enfermedad tiroidea y otros 4 de diversas enfermedades autoinmunes. Entre la patología asociada que presentan los pacientes, destacan: déficit de IgA (2), déficit de IgA + IgM (1), s. de Turner (1), vitíligo (1), lupus (1) y PTI (1). Los síntomas más frecuentes entre los 18 pacientes son: bocio (16), taquicardia (13), inquietud-nerviosismo (11), temblor fino (9), pérdida de peso (8) y sudoración (6). En 5 casos se realizó gammagrafía tiroidea y en todos los pacientes se llevó a cabo el estudio y seguimiento ecográfico; 2 pacientes presentan un bocio multinodular y los 16 restantes un bocio difuso con ecogenicidad homogénea o heterogénea. En todos los pacientes se constató analíticamente la hiperfunción tiroidea. En 15/17 los anticuerpos antitiroperoxidasa fueron positivos y en 11/15 lo fueron los anticuerpos antirreceptor de TSH. Un paciente, varón, diagnosticado a los 2,1 años de edad, con autoanticuerpos negativos, historia prolongada de sintomatología sugestiva de hipertiroidismo, edad ósea de 5,5 años y antecedentes de padre hipertiroideo con anticuerpos anti-receptor de TSH negativos, parece tratarse de un caso de hipertiroidismo no inmune. El tratamiento inicial fue farmacológico en todos los pacientes, utilizándose el carbimazol a 0,3-0,5 mg/kg/día, hasta 1 mg/kg/día en un paciente; 3 pacientes precisaron propranolol y 1 corticoides. Un total de 13 pacientes requirieron tratamiento con l-tiroxina durante el tratamiento antitiroideo. En 2 pacientes, tras recidivar el proceso, se recurrió a la tiroidectomía y en 1 al tratamiento con 131I.
Conclusiones: La EG es poco frecuente en la edad pediátrica, siendo la principal causa de hipertiroidismo durante la misma. Se asocia a otros procesos autoinmunes. La principal opción terapéutica es la farmacológica, pero cada vez se usan más el 131I y la cirugía, también en la edad pediátrica. En los casos con anticuerpos negativos debería pensarse en la posibilidad de una etiología no inmune.
Oftalmopatía tiroidea atípica. A propósito de un caso
M.A. Fuentes Castello, S. de Murcia, F. Revert y F. Vargas
Pediatría. Hospital Universitario de Elche. España.
Introducción: La oftalmopatía de Graves, la más frecuente manifestación extratiroidea de la enfermedad de Graves, pese al largo tiempo transcurrido desde su primera descripción, sigue siendo un enigma en cuanto a su etiopatogenia y un dilema en cuanto a su tratamiento. Aunque la mayoría de veces se asocia al hipertiroidismo de Graves, en ocasiones se ha descrito asociada a tiroiditis de Hashimoto o incluso a pacientes que permanecen eutiroideos, tanto en el momento de la oftalmopatía como antes o después de ésta. Situación ésta última descrita desde hace más de 40 años y conocida como "enfermedad de Graves eutiroidea". El caso que a continuación presentamos es una oftalmopatía que va más allá en lo atípico de su presentación clínica, ya que además de eutiroidea es, pese a ser muy llamativa, unilateral.
Caso clínico: Niña de 10,9 años remitida a nuestra consulta desde Oftalmología por exoftalmos unilateral izquierdo, progresivo, de 3 meses de evolución e inicio relativamente agudo, sin otra sintomatología asociada. Presenta la siguiente analítica: T4 libre de 2,2 ng/dl (0,8-2) y TSH de 0,011 μUI/l (0,6-4) sin síntoma alguno de hipertiroidismo. Como antecedentes destacan un vitíligo y una celiaquía, diagnosticada a los 13 m de edad. En la exploración física destaca una proptosis severa del globo ocular izquierdo (24 mm) y retracción del párpado superior, sin dolor ni otros signos inflamatorios. No diplopía ni pérdida de la agudeza visual con examen funduscópico normal. No asocia bocio ni otros hallazgos patológicos. Ya realizadas ecografía y TC orbitarios que resultaron normales, solicitamos nueva analítica con T4 libre de 1,9 ng/dl, TSH de 0,006 μUI/l, anticuerpos antirreceptor de TSH de 17 UI/ml (positivos a partir de 14) y anti-TPO de 30,5 (normal < 40). La gammagrafía tiroidea muestra una hipercaptación tiroidea leve (9 %) sin otros hallazgos. Ante la persistencia del exoftalmos y la ausencia de un claro patrón analítico y clínico sugerente de una enfermedad de Graves típica, solicitamos una RNM orbitaria en la que se informa de una leve hipertrofia del músculo recto superior izquierdo sin efecto masa en su interior con el resto de estructuras intraorbitarias normales. Tras dicho resultado se nos plantea el diagnóstico diferencial de una miositis orbitaria idiopática, que afecta a músculos aislados de la musculatura ocular extrínseca, versus una oftalmopatía de Graves atípica (por ser unilateral y por asociar hipertiroidismo sólo subclínico, tal y como se describe en algunos casos). Decidimos iniciar tratamiento esteroideo (2 meses de prednisona oral a 1,5 mg/kg/día, con retirada a lo largo de 4 meses), el cual se recomienda en ambas entidades caso de afectación severa como en nuestra paciente. Por fortuna y pese a haber desarrollado un síndrome de Cushing transitorio como efecto colateral, se objetivó una mejoría progresiva desde del inicio del tratamiento hasta la desaparición casi completa de la proptosis. En los casi 4 años de seguimiento realizados, no ha desarrollado recidiva del exoftalmos ni hipertiroidismo ni elevación de los anticuerpos antirreceptor de TSH.
Comentarios: Planteamos como diagnóstico más probable la oftalmopatía tiroidea atípica en perjuicio de la mencionada miositis, ante la difícil correlación causal entre una discreta asimetría en el músculo orbitario citado, por lo demás indoloro, y un exoftalmos tan severo. Resulta de vital importancia el diagnóstico diferencial de estas entidades con otras como la tumoral ante las diferentes implicaciones pronósticas y terapéuticas que conlleva. El diagnóstico de una oftalmopatía tiroidea en pacientes eutiroideos es difícil sin poderse establecer con pruebas objetivas, dependiendo sólo para ello del exámen clínico. Aunque los TBII (TSH binding inhibitor immunoglobulins) pueden bloquear la síntesis hormonal tiroidea y explicar así oftalmopatías asociadas a estados hipotiroideos o eutiroideos, lo cierto es que en el 66 % de estas oftalmopatías eutiroideas dichos Acs TBI son negativos. De hecho, se ha descrito que el Ac estimulador del receptor de TSH o TSAb (thyroid stimulating antibody) se asocia al 80 % de los casos de oftalmopatía eutiroidea, siendo (al revés de lo que parecería más lógico) mucho más sensible que TBII, por tanto, para su diagnóstico.
Bocio endémico en un área rural de la Comunidad Valenciana
B. Peris Roig 1, N. Atienzar Herráez 1, F. Calvo Rigual 1, A. Merchante Alfaro 1, S. Selfa Moreno 1, J.M. Tenía Burillo 1 y M.J. López García 2
Servicio de Pediatría. 1Hospital Lluís Alcanyís de Xàtiva. 2Hospital Clínico Universitario de Valencia. Xàtiva. Valencia. España.
Introducción: España está incluida por el ICCIDD dentro de los países europeos con déficit de yodo. Una alta prevalencia de bocio existe en zonas deficitarias de yodo. Interesa valorar cuál es la situación actual en nuestro medio.
Material y métodos: Estudio epidemiológico, descriptivo, transversal, estratificado por sexo y edad en un área rural de la Comunidad Valenciana. Población estudiada: niños de 6 a 14 años de 20 centros escolares con un muestreo aleatorio de 55 niños en cada uno.
Método de estudio: Determinación de la existencia de bocio mediante palpación e inspección del tiroides por dos examinadores independientes. Clasificación según criterios de la PAHO y OMS (modificado por Querido): se considera bocio la presencia de estadio 0B o mayor. Determinación de la yoduria en muestra de orina casual recogida en la escuela (método de Benotti y Benotti). Recogida de datos de filiación y antropométricos y realización de encuesta nutricional de yodo. Estudio hormonal tiroideo y anticuerpos antitiroideos en los niños con bocio.
Resultados: El tamaño muestral fue de 1.082 (102 no respondieron y 52 estuvieron ausentes). Se estudiaron 928 niños (478 varones y 450 mujeres) con una edad media de 9,4 + 2,2 años. Se encontró bocio en 313 (33,7 %) (IC 30,7-36,9). De ellos, 195 estaban en estadio 0B, 112, en estadio I, y 6, en estadio II. No hay diferencias en cuanto a sexo. Elevada concordancia entre los dos examinadores (índice de Kappa 0,83). Se encontró una correlación inversamente proporcional entre prevalencia de bocio y el nivel socioeconómico. La mediana de yoduria fue de 155 mg/l, lo que haría suponer una ingesta adecuada de yodo. No había correlación entre yoduria y presencia o no de bocio. Se diagnosticaron 13 casos con tiroiditis autoinmune, 18 con hipotirodismo subclínico y 1 caso con hipertirodismo subclínico entre los niños con bocio.
Conclusiones: Se detectó una endemia bociosa con niveles de yoduria normales, lo cual podría interpretarse como el reflejo de una fase de transición hacia una mejoría del déficit de yodo en la zona estudiada. La patología autoinmune como causante de bocio únicamente explicaría un 4 % de los casos.
Enfermedad de Graves Basedow. Revisión de 12 casos
M. Torrabías Roda, R. Nosas, V. Aldecoa, R. Corripio, M. Fletas y A. Pérez
Unidad de Endocrinología Pediátrica. Consorci Hospitalari Parc Taulí. Barcelona. España.
Objetivos: La enfermedad de Graves Basedow es la causa más frecuente de hipertiroidismo en pediatría. Analizaremos los casos diagnosticados en nuestra Unidad.
Material y métodos: Revisión retrospectiva de las historias clínicas de los pacientes menores de 15 años diagnosticados de enfermedad de Graves Basedow entre los años 1992 y 2004 en nuestro centro.
Resultados: Se han revisado 12 casos. La mayoría (91 %) son niñas. La edad al diagnóstico fue de 9 años (rango 7-14 años). Existían antecedentes familiares de patología tiroidea en el 58 % de los casos. Los síntomas más frecuentes fueron: aumento de ingesta sin ganancia de peso (42 %), insom nio (42 %), nerviosismo (33 %), labilidad emocional (33 %), intolerancia al calor (33 %) y temblores (33 %). Los signos encontrados fueron: bocio (91 %), taquicardia (67 %) y retracción palpebral (58 %). Todos los pacientes presentaron anticuerpos estimuladores del tiroides al inicio de la enfermedad o durante su evolución. En todos la ecografía tiroidea mostraba aumento de tamaño de la glándula tiroidea y de su vascularización en el estudio Doppler. Todos los casos recibieron inicialmente tratamiento médico antitiroideo y se logró la remisión en dos casos (16 %); un paciente desarrolló leucopenia como efecto adverso. En tres casos (25 %) con recaídas se recurrió a radioyodo consiguiéndose la curación; en 2 pacientes se originó hipotiroidismo residual. El 59 % restante se mantienen eutiroideos en la actualidad con tratamiento frenador. En ningún caso se practicó cirugía.
Comentarios: La remisión definitiva de la enfermedad de Graves-Basedow con tratamiento médico es poco habitual, aunque en nuestro medio se considera la primera opción terapéutica. Debido a las recaídas frecuentes de los pacientes con dicho tratamiento a menudo hay que recurrir a la ablación de la glándula tiroidea, ya sea quirúrgica o por radioyodo, aunque se origine un hipotiroidismo secundario que resulta de más fácil manejo.
METABOLISMO-NUTRICION
Repercusión del sobrepeso y obesidad en la calidad de vida de niños y adolescentes
I. Riaño Galán 1, P. Santos Rodríguez 1, M. Fernández-Fidalgo 2, F. Rivas Crespo 3 y J.A. Fernández-López 4
1Servicio de Pediatría. Hospital Carmen y Severo Ochoa. 2Facultad de Psicología. Universidad de Oviedo. 3Hospital Universitario Central de Asturias. 4Centro de Salud de Riosa. Asturias. España.
Introducción: La obesidad en la infancia es un grave problema de salud pública de importancia creciente por las consecuencias a corto y largo plazo sobre la salud. Los adolescentes con obesidad o sobrepeso presentan problemas de comportamiento y baja autoestima.
Objetivo: Evaluar la repercusión del sobrepeso y obesidad en la calidad de vida relacionada con la salud (CVRS) de niños y adolescentes del ámbito rural.
Sujetos y métodos: Estudio transversal de los 150 niños y adolescentes de dos colegios públicos del área suroccidental de Asturias, con aplicación del cuestionario Kindl, en su versión adaptada y validada al español. Se autoadministró el módulo de 8-12 años (Kid-Kindl) a 66 niños (32 varones) y el módulo de 13-16 años (Kiddo-Kindl) a 84 adolescentes (43 varones). Dicho cuestionario consta de 24 preguntas agregadas a 6 dimensiones básicas de 4 ítems cada una: bienestar físico, bienestar emocional, autoestima, familia, amigos, escuela. Las puntuaciones obtenidas a partir de las medias de cada dimensión se transformaron a una escala de 0-100, donde una puntuación mayor representa una mejor CVRS. También se ha obtenido un índice global de CVRS. Además, se recogieron datos somatométricos: peso, talla, índice de masa corporal (IMC) (kg/m 2), calificándolos en desviaciones estándar para edad y sexo según las gráficas de la Fundación Orbegozo. Según el IMC, los pacientes se clasifican como normopeso (< P85), sobrepeso (P85-P95) u obesidad (> P95).
Resultados: El 6,1 % de los niños del grupo de 8 a 12 años presenta sobrepeso y el 27,7 %, obesidad. El sobrepeso es del 5 % y la obesidad del 31 % en el grupo de 13-16 años. Las puntuaciones finales de CVRS son más bajas, en todas las dimensiones, independientemente del peso, en los adolescentes. En el grupo de 8-12 años las puntuaciones de CVRS de los obesos son superiores globalmente y en todas las dimensiones a las de los sanos, si bien las diferencias no alcanzan significación estadística. Sin embargo, los obesos adolescentes presentan peores puntuaciones de CVRS que los adolescentes con peso normal en bienestar emocional (80,2 frente a 81,0) y autoestima (61,6 frente a 66,3) si bien tampoco alcanzan significación estadística. La autoestima se encontró menor entre las niñas en ambos grupos de edad (70,9 frente a 81,4 en el grupo de 8-12 años; 58,9 frente a 70,1 en el grupo de 13-16 años, p < 0,05).
Conclusiones:1) La prevalencia de sobrepeso y obesidad en niños y adolescentes de medio rural estudiado es alta, incrementándose con la edad. 2) En el grupo de niños de 8-12 años el sobrepeso y obesidad se acompaña de mayores puntuaciones de CVRS, con un sentimiento de bienestar general en todos los ámbitos (gordos felices). 3) Por el contrario, entre los adolescentes la obesidad ya provoca un deterioro del bienestar psíquico y del sentimiento de autoestima, en relación con sus iguales sanos. Es importante introducir la valoración de la calidad de vida en la asistencia de los adolescentes, desde una visión psicosocial. Ampliar el número de casos estudiados nos permitirá corroborar estos aspectos de evidente relevancia asistencial.
Enfermedad de hígado graso no alcohólica en el niño obeso
J.M. Fernández García, A. Goicoechea, E. Ocete, T. Rebora, M. Garofano, C. Pérez Ballesteros, J. Salmeron y A. Ruiz-Extremera
Servicio de Endocrinología Infantil e Intensivos. Hospital Clínico Universitario San Cecilio. Granada. España.
Objetivo: Conocer la prevalencia de esteatosis hepática (ecografía) y de aumento de ALT como expresión de enfermedad de hígado graso no alcohólico en la población obesa infantil en España, así como posibles factores implicados en esta asociación.
Material y método: Se estudian 76 pacientes pediátricos remitidos a la consulta de endocrinología para estudio por sobrepeso-obesidad y se clasifican como sobrepeso cuando presentan un índice de masa corporal (IMC) mayor del percentil 90 y como obesidad si el IMC > P97, según las Curvas de Referencia para la Tipificación Ponderal de la población infantil y juvenil del grupo colaborativo AEP-SENC-SEEDO. Se realiza ecografía hepática para definir el grado de esteatosis. Tras un ayuno de 10 h se toman muestras de sangre para determinación de aminotransferasas, glucosa, insulina, péptido C, triglicéridos, colesterol, bilirrubina, albúmina, ferritina. Se congelan 2 ml de suero para estudio de los marcadores de estrés oxidativo. Se determina la prevalencia de hígado graso en la población obesa infantil en nuestro medio y la actividad sérica de ALT, y se relaciona con los marcadores metabólicos que explican esta asociación (resistencia a la insulina y marcadores de estrés oxidativo). La resistencia a la insulina se determina por el método HOMA. El análisis estadístico se efectúa mediante el programa SSPS10.
Resultados: El 43 % de los obesos presentaban algún grado de esteatosis en la ecografía sin que existiera una diferencia significativa entre ambos sexos. De los niños con sobrepeso ninguno presentó esteatosis. La actividad sérica de ALT estuvo elevada en el 7,41 % de los niños. Todos los niños con actividad ALT alta tenían un IMC > P97 y presentaban esteatosis. El 24,2 % de los niños tenía un índice HOMA > 3,8, siendo este índice significativamente más alto en los obesos con esteatosis. Algunos marcadores de estrés oxidativo también dieron valores significativamente más altos en el grupo con esteatosis.
Conclusión: Este estudio preliminar señala que existe una elevada prevalencia de hígado graso en la población obesa infantil de nuestro medio. La ecografía hepática es un excelente instrumento para detectar la esteatosis. La resistencia a la insulina está aumentada en los pacientes con esteatosis. Esta resistencia aumentada y el aumento de algunos marcadores de estrés oxidativo pueden estar implicados en el desarrollo de esteatohepatitis con sus graves consecuencias aunque hace falta completar el estudio para definirlo con precisión.
Índice de masa corporal y porcentaje de masa grasa en el diagnóstico de obesidad en niños y adolescentes: evaluación mediante Curvas Receiver-Operating Characteristics
C. Azcona Sanjulián y S. Aguilera Albesa
Unidad de Endocrinología Pediátrica. Clínica Universitaria. Universidad de Navarra. Pamplona. España.
Introducción: El índice de masa corporal (IMC) se utiliza para diagnosticar la obesidad en niños y adolescentes. Sin embargo, en algunos casos puede infradiagnosticar esta enfermedad.
Objetivos: El propósito de este estudio fue evaluar la capacidad del IMC para identificar correctamente a los pacientes obesos (clasificados según el porcentaje de masa grasa).
Material y métodos: El porcentaje de masa grasa (MG) se determinó mediante pletismografía por desplazamiento de aire (sistema BOD-POD, Life Measurements Instruments, Concord, California, EE.UU. software versión 1,69) en 263 (rango de edad: 5 a 22 años) niños y adolescentes caucasianos (148 mujeres y 115 hombres). Se consideró obeso a un paciente cuando su porcentaje de MG era mayor del 25 % en niños y del 30 % en niñas. El IMC-SDS se calculó según estándares españoles. Se utilizó el test de McNemar para comparar las frecuencias de obesidad. Las curvas Receiver Operating Characteristics (ROC) se obtuvieron contrastando sensibilidad versus 1-especificidad para obtener el umbral de decisión y determinar así el punto de corte óptimo de IMC que defina obesidad, tomando el porcentaje de MG como referencia.
Resultados: La frecuencia de obesidad medida por el porcentaje de MG (42,2 %) fue significativamente (p < 0,NT FACE="Symbol" SIZE=2>> 2 SDS) (22,8 %). La definición actual de obesidad basada en un IMC mayor de 2SDS obtuvo una sensibilidad del 50 % (IC 95 %: 37,1 a 56,7) y una especificidad del 97 % (IC 95 %: 83,96 a 97,3). El área bajo la curva ROC era de 0,92, dato que indica una máxima eficacia diagnóstica. Con el análisis de las curvas ROC, el punto de corte sugerido (índice máximo de Youden) para el IMC fue 1,12 SDS. En este punto de corte para el IMC, la sensibilidad y especificidad fueron de 82,0 % (IC 95 %: 69,5 a 86) y 88,8 % (IC 95 %: 77,8 a 90,72), respectivamente, con un valor predictivo positivo y negativo de 65,4 y 95,1 %, respectivamente (asumiendo una prevalencia de obesidad en la población del 20 %).
Conclusiones: El punto de corte óptimo que define la obesidad según el IMC en este grupo de pacientes, es menor que el utilizado habitualmente para su diagnóstico. Una nueva definición de obesidad tomando un punto de corte de IMC más bajo, o basado en el porcentaje de MG, nos llevará a una mejora en la correcta identificación de niños y adolescentes con obesidad y a una mejor aplicación de las pautas de alimentación y de estilos de vida.
Efectos de la dieta mediterránea sobre el perfil lipídico en pacientes prepuberales con obesidad
A.M. Lechuga Sancho y J.L. Lechuga Campoy
Servicio de Pediatría. Sección Endocrinología Pediátrica. Hospital Universitario Puerta del Mar. Cádiz. España.
Introducción: En los últimos 15 años se ha duplicado la incidencia de obesidad infantil en España, que ocupa así el cuarto puesto en la clasificación europea. Las complicaciones cardiovasculares que asocia en la edad adulta comienzan ya en la infancia, por lo que la intervención dietética y la adquisición de unos hábitos de vida cardiosaludables deben comenzarse entonces.
Objetivos: Puesto que la dieta mediterránea es hipocalórica con respecto a otras alimentaciones y rica en ácidos grasos insaturados, el objetivo de este estudio es analizar los efectos sobre el perfil lipídico de la dieta mediterránea en una población de niños prepuberales con obesidad.
Pacientes y métodos: Se reclutaron 70 pacientes (44 niños de edades comprendidas entre 5,5 y 12,7 años, y 26 niñas de 4,99 a 10,6), enviados por sus pediatras de atención primaria por obesidad. Se realizó analítica basal y se indicaron a los pacientes y familiares las recomendaciones higiénico-dietéticas que seguir. Se realizó control clínico mensual y analítico al cabo de los 6 meses. Quedaron excluidos del estudio los pacientes que comenzaron la pubertad durante el seguimiento y aquéllos con obesidad orgánica.
Resultados: El 37,14 % (n = 26) de los pacientes lograron una disminución de peso superior o igual a dos desviaciones estándar para la edad y sexo, mientras que el 62,86 % restante no logró una disminución significativa. La evolución del perfil lipídico se realizó comparando entre estos dos grupos. Las concentraciones de triglicéridos, colesterol total y colesterol-LDL disminuyeron en el grupo respondedor, aunque sin significanción estadística. Las concentraciones de colesterol-HDL tras 6 meses de tratamiento, incrementaron significativamente (p < 0,05) en los pacientes que lograron la disminución de peso.
Conclusiones: El tratamiento con dieta mediterránea durante 6 meses logra una mejora significativa del perfil lipídico de los pacientes prepuberales con obesidad que pierden peso, fundamentalmente a expensas de incrementar el colesterol-HDL, logrando así una disminución del riesgo cardiovascular en los mismos.
Esteatosis hepática e insulinorresistencia en población obesa pediátrica
M. López Capapé, E. Colino, C. Mustieles, L. Golmayo, J. Corbatón, R. Barrio y M. Alonso
Servicio de Pediatría. Unidad de Endocrinología Pediátrica. Hospital Universitario Ramón y Cajal. Madrid. España.
Introducción: La esteatosis hepática no alcohólica (EHNA) puede representar una manifestación del síndrome metabólico y constituye un amplio espectro clínico-patológico (esteatosis, esteatohepatitis, fibrosis, cirrosis hepática). La EHNA, pero más específicamente la esteatohepatitis, se asocia a insulinorresistencia y su incidencia está aumentando en la edad pediátrica de forma paralela a la obesidad.
Objetivo: Analizar la prevalencia de EHNA, identificada por ecografía, en nuestra población pediátrica obesa. Describir las características clínicas y bioquímicas de la EHNA, así como su asociación a insulinorresistencia y otros parámetros de síndrome metabólico.
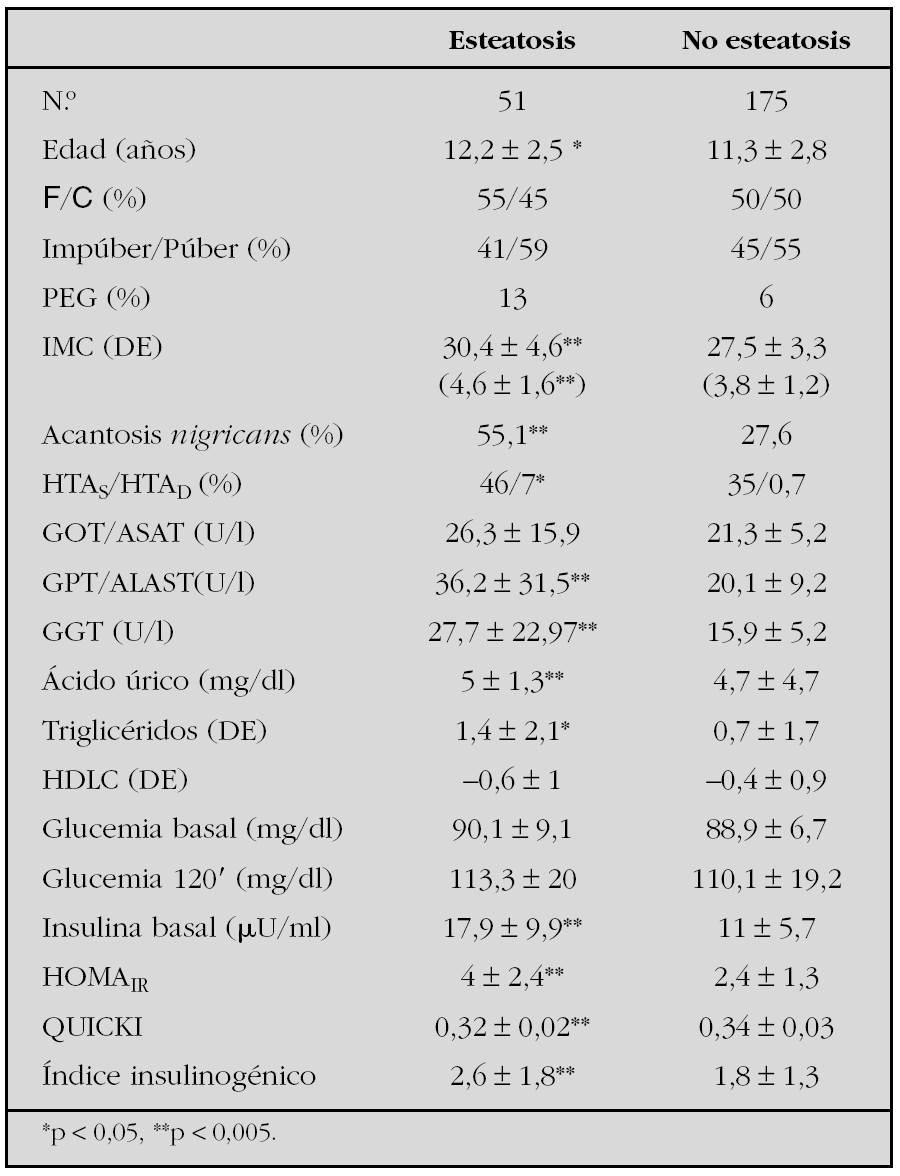
Métodos: Se estudiaron 226 niños con obesidad (IMC > 2 DE), con edades comprendidas entre 4 y 17 años. Se valoraron sus antecedentes, la existencia de acantosis e HTA en la exploración y se les realizó ecografía, sobrecarga oral de glucosa (SOG), perfil lipídico, hepático y ácido úrico en ayunas. Se definió EHNA según los hallazgos ecográficos (hiperecogenicidad hepática) habiendo excluido otras causas de daño hepático. Se comparó el grupo con esteato sis frente al grupo de pacientes sin esteatosis. La insulinorresistencia, insulinosensibilidad y secreción de célula b fueron estimados con HOMAIR, QUICKI e índice insulinogénico, respectivamente. Se ajustó el IMC por edad y sexo según tablas de Hernández y los lípidos y lipoproteínas según tablas del estudio de Fuenlabrada para población española. Se definió HTA como TA ≥ P90 según la Task Force. Los resultados se expresan en media ± DE o %. La comparación entre grupos se realizó con t-Student y test no paramétricos. Se consideró nivel de significación p < 0,05.
Resultados: Se detectó esteatosis en 51 pacientes (22 %). La tabla 1 resume las características clínicas y bioquímicas de los pacientes. El 23 % de los pacientes con esteatosis presentó alteración enzimática (GPT ≥ 40U/l) frente al 5 % del grupo sin esteatosis (p < 0,001). Se detectó en el grupo con esteatosis un 4 % de ATG y un 4 % de AGA (ADA 1997).
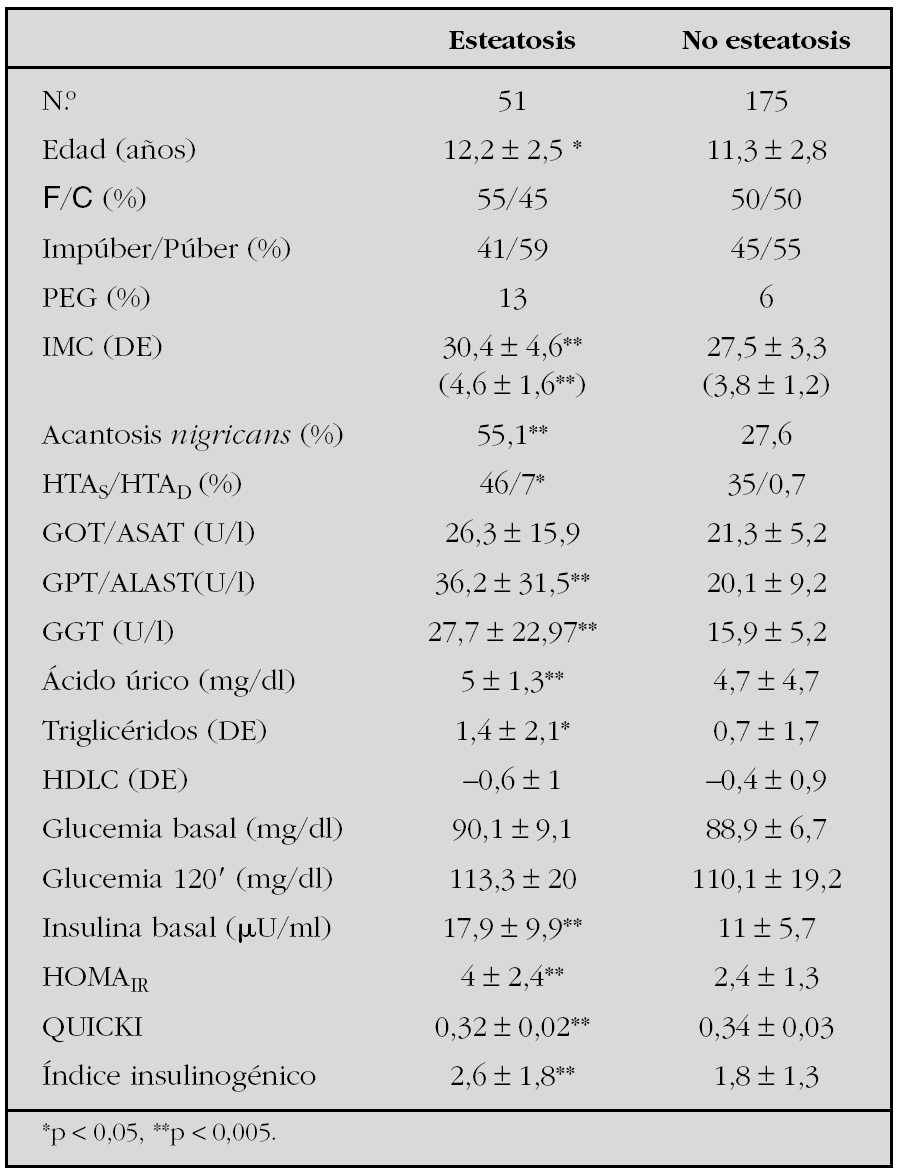
Conclusiones: La insulinorresistencia parece tener una gran influencia en la patogenia del hígado graso. El grado de obesidad está significativamente asociado a EHNA.
La grelina posprandial está alterada en niños obesos prepuberales
M. Gil Campos, J. Linde Gutiérrez, M.C. Ramírez Tortosa, R. Cañete Estrada y A. Gil Hernández
Servicio de Endocrinología Pediátrica. Hospital Universitario Reina Sofía. Córdoba. Departamento de Bioquímica y Biología Molecular. Córdoba. España.
Objetivo: Este trabajo pretende evaluar las concentraciones plasmáticos de grelina posprandiales en niños obesos y en un grupo control en estado prepúber.
Materiales y métodos: Se seleccionó un grupo de 34 niños obesos (O) y otro de 20 niños control (C) entre 6 y 13 años, ambos en estado prepuberal, apareados por edad y sexo. Se realizaron medidas antropométricas para su clasificación. En los niños obesos, el índice de masa corporal (IMC) fue de 29, con un percentil mayor de 97. El estado prepuberal se estableció mediante criterios clínicos (estadios de Tanner) y medidas hormonales. Los pacientes recibieron un desayuno de composición nutricional previamente fijada (428 kcal). Se extrajeron muestras de sangre en ayunas, a la hora, 2 h y a las 3 h de la ingesta del desayuno. Las concentraciones de grelina se determinaron mediante radioinmunoensayo. Los análisis estadísticos se llevaron a cabo utilizando el programa informático SPSS 11,5. Para evaluar los efectos de obesidad y tiempo posprandial se utilizó el modelo lineal general de ANOVA para medidas repetidas.
Resultados: Los niveles de grelina en ayunas fueron similares en el grupo de obesos y en el control, y descendieron tras la primera hora de la ingesta del desayuno estandarizado (P = 0,007). A las 2 h, las concentraciones de grelina se mantuvieron cercanas a las de la primera hora. En el grupo de obesos, los niveles de grelina retornaron a los basales tras la tercera hora (P < 0,05).
Conclusiones: Tras la ingesta de un desayuno estandarizado, los niños obesos recuperan los niveles de grelina basales más rápido que el grupo control. Este comportamiento de la grelina puede tener un gran impacto en la dieta favoreciendo una mayor ingesta en los niños obesos.
Estudio financiado por el Proyecto FIS 020826 y la Fundación Salud 2000.
Cambios en la actividad lipolítica y en el metabolismo hepático de ácidos grasos en niños obeso: relación con la presencia de acantosisnigricans
B. Bonet Serra, I. Sánchez-Vera, A. Quintanar Rioja, M. Viana Arribas, J. Pérez-Lescure y M. Bueno Campaña
Servicio de Pediatría y Neonatología. Fundación Hospital Alcorcón. Universidad San Pablo-CEU. Madrid. España.
Introducción: La insulina es un potente inhibidor de la lipolisis, por lo que en situaciones de resistencia a la insulina es de prever que la lipólisis se active, aumentando las concentraciones plasmáticas de glicerol y de ácidos grasos libres (AGL), así como de los productos derivados de su metabolismo hepático.
Objetivo: determinar en niños obesos con y sin acantosis nigricans (AN), proceso asociado a la resistencia a la insulina, diferentes parámetros relacionados con la lipólisis (ácidos grasos libres y glicerol) y el metabolismo hepático de los ácidos grasos: beta-hidroxibutirato (β -HO-but) y triglicéridos (TG).
Diseño experimental: En su primera visita a la consulta de endocrinología pediátrica se estudiaron 101 niños, 74 obesos, índice de masa corporal (IMC) superior al percentil 97 para su edad y sexo), de los cuales 30 presentaron acantosis nigricans. También se estudiaron 27 pacientes que no presentaron patología y no eran obesos. La distribución por edad, grado de desarrollo puberal y sexos fue similar entre los tres grupos. En todos los pacientes se obtuvieron muestras de sangre tras 12 h de ayuno y se determinaron los valores plasmáticos de AGL, glicerol, TG, β -HO-but, glucosa e insulina. También se calculó el índice de resistencia a la insulina (IRI).
Resultados: Los niños obesos con AN presentaron un IMC de 30,8 kg/m 2 superior al observado en los obesos sin AN que fue de 27,5 kg/m 2 (p < 0,001) y del grupo control (IMC: 18,0 kg/m 2; p < 0,001) (tabla 1).

Cuando se llevó a cabo una regresión lineal múltiple se observó que las concentraciones plasmáticas de triglicéridos dependían tanto del IMC como de la presencia de AN, indicando que la presencia de AN es un factor independiente del grado de obesidad. Como cabría esperar, el IRI fue mayor en los niños obesos con AN que en los OB-AN y el grupo control.
Conclusiones: En niños obesos con AN el metabolismo lipídico no se ve afectado por la resistencia a la insulina, como pone de manifiesto la menor actividad lipolítica, la menor síntesis de β -hidroxibutirato y la elevación de los triglicéridos plasmáticos. Este estado de hiperinsulinismo sin resistencia a la insulina, en cuanto al metabolismo lipídico se refiere, favorecería el acumulo de grasas y, por lo tanto, la obesidad.
Metformina en niños y adolescentes con ganancia ponderal excesiva secundaria a psicofármacos
C. Azcona Sanjulián y A. Díez Suárez
Unidad de Endocrinología Pediátrica. Clínica Universitaria. Universidad de Navarra. Pamplona. España.
Introducción: El aumento de peso se ha descrito como un efecto adverso asociado al tratamiento prolongado con psicofármacos hasta en un 50 % de los pacientes. Para disminuir esta ganancia ponderal, se han utilizado metformina, topiramato o reboxetina. Recientemente se ha aprobado el empleo de metformina en niños. Este fármaco produce un aumento del metabolismo mitocondrial y estimula la β -oxidación mitocondrial, lo que se traduce en una disminución del peso.
Método: Realizamos un análisis retrospectivo de los pacientes tratados con metformina por aumento de peso secundario a psicofármacos, entre los años 2000-2004. El análisis estadístico se realiza con el programa SPSS, versión 11.0. Los datos se expresan en mediana y amplitud intercuartil (AIC).
Resultados: Se indicó tratamiento con metformina a 9 pacientes. Uno de ellos es eliminado del estudio por falta de seguimiento. Los 8 pacientes analizados, 5 mujeres y 3 varones, tenían una mediana de edad de 15,7 (3,2) años. Los diagnósticos psiquiátricos fueron: 5 pacientes (62,5 %) con trastorno bipolar, 1 (12,5 %) con trastorno del humor no especificado, 1 (12,5 %) con trastorno conversivo y 1 (12,5 %) con trastorno orgánico de la personalidad asociado a esclerosis tuberosa. Los psicofármacos a los que se asoció la ganancia ponderal secundaria eran ácido valproico y olanzapina. Todos los pacientes reconocían aumento del apetito y desestructuración en los hábitos alimentarios desde el inicio de esta medicación. El IMC-SDS antes de iniciar el tratamiento con psicofármacos fue de 1,15 (1,58), que aumentó a 3,74 (3,65). Tras la administración de metformina durante 11 meses (9,75), disminuyó a 2,41 (3,36) (p = 0,125). La pérdida media de peso fue de 5,77 kg (14,1) (p = 0,284). La dosis de metformina fue 1.912 mg (1.275). Un paciente presentó diarrea y otro hipoglucemia.
Conclusiones: La metformina es un fármaco que puede ser eficaz y es bien tolerado en niños y adolescentes para la prevención y tratamiento del aumento de peso asociado al uso prolongado de psicofármacos.
Obesidad endocrina: enfermedad de Cushing en un niño de 11 años
J. Ferragut Martí 1, M. Caimari Jaume 1, A. Mas Bonet 2, M. Herrera Savall 2 y B. Oliver Abadal 3
Servicio de 1Pediatría y 2Radiología. Hospital Universitario Son Dureta. Palma de Mallorca. Baleares. 3Neurocirugía. Hospital Mútua de Terrassa. Barcelona. España.
Introducción: La obesidad infantil raramente se debe a una causa endocrina identificable. Aunque la gran mayoría de casos son causados por una ingesta calórica excesiva, hay que prestar una especial atención a la velocidad de crecimiento del paciente, a fin de descartar un origen endocrino.
Caso clínico: Varón de 11 años que consulta en noviembre de 2002 por aumento de peso gradual en los últimos 3 años. Se comprobó que, paralelamente, había disminuido la velocidad de crecimiento, pasando de P40 a P4 en los últimos 3 años. Apetito referido como normal. Embarazo y parto normales. Peso al nacer 3.250 g. Longitud 49 cm. Período neonatal normal. Desarrollo psicomotor normal. Padre, 43 años, mide 168 cm. Madre 41 años, 154 cm. Hermano 5 años, sano. Exploración: Peso 43.100 g (+ 1,02 SDS); talla 133,1 cm (1,7 SDS); PA 129/88; pulso 90/min. Obesidad de predominio en cara y cuello, que es corto, doble relieve en mentón. No hirsutismo. Auscultación cardiopulmonar normal. Abdomen normal. Genitales normales. G1. P1-2. Testes 4 ml. No vello axilar. Fondo de ojo normal. La analítica de rutina, incluyendo hemograma, glucemia, urea, creatinina, ácido úrico, colesterol, AST, ALT, proteínas totales, fosfatasa alcalina, calcio, fósforo, sodio, potasio y cloro fue normal. FT4 1,03 ng/dl. TSH 1,15 μIU/ml. Prolactina 8,43 ng/ml. Cortisol 27,15 μg/dl. Δ4-A 2,9 ng/ml. Testosterona 0,44 ng/ml. ACTH 22 pg/ml. Cortisol urinario 363,6 μg/24 h, postextracción 259,2 μg/24 h. Ritmo circadiano de cortisol: 8 h 22,72 μg/dl, 20 h 30,42 μg/dl. Test de supresión con dexametasona (DXM): cortisol basal 22,72 μg/dl, post-DXM 20,45 μg/dl. Test de CRH tras supresión con 6 mg de DXM a las 23 h: cortisol basal 3,43 μg/dl, pico máximo 3,77 μg/dl, ACTH basal 13,1 pg/ml, pico máximo 16,6 pg/ml. TC de abdomen normal. TC hipofisario: área densa redondeada en la porción anterosuperior de la hipófisis. RM hipofisaria: lesión lobulada, nodular, de 8 mm de diámetro, en la región supraselar, margen anterior de la hipófisis y espacio prequiasmático; el resto de la glándula aparece ligeramente abombada en la superficie anterior. El tallo hipofisario, nervios ópticos y quiasma son normales. Son hallazgos compatibles con microadenoma hipofisario localizado en la línea media del lóbulo anterior de la hipófisis, con extensión hacia la cisterna supraselar y región prequiasmática. El paciente fue intervenido, previa administración de ketoconazol 200 mg/12 h. Tras la intervención no se modificó la situación clínica y se mantuvieron concentraciones elevadas de cortisol plasmático (29,67 μg/dl) y en orina (409,5 μg/24 h). La RM hipofisaria seguía mostrando la imagen del adenoma intraselar y extraselar. En febrero de 2004 se practicó hipofisectomía subtotal. La respuesta ha sido favorable, normalizándose las cifras de cortisol en sangre y orina. Lleva tratamiento sustitutivo con GH, tiroxina e hidrocortisona. Su aspecto ha cambiado, y en los últimos 7 meses ha crecido 7 cm y ha perdido 7600 g. Ha iniciado el desarrollo puberal (P2, testes 5 ml) y permanece asintomático. La RM hipofisaria muestra una silla turca parcialmente vacía, con restos glandulares intraselares, se visualiza la neurohipófisis.
Discusión: La obesidad se ha convertido en los últimos años en la causa más frecuente de consulta en endocrinología pediátrica, aunque sólo excepcionalmente es debida a causas hormonales. La mayoría de veces se debe a ingesta calórica excesiva, unida al sedentarismo (como ver televisión en exceso) y a una determinada predisposición genética. Se asocia a algunos síndromes hereditarios infrecuentes, como los de Laurence-Moon-Biedl-Bardet, Alström y Prader-Willi. Es también un signo acompañante de algunas enfermedades endocrinas (hipotiroidismo e hipercortisolismo en especial), y en estos casos es esencial el diagnóstico precoz, ya que es posible el tratamiento. La clave esencial para el diagnóstico es la cuidadosa observación de la velocidad de crecimiento. Los niños con obesidad simple son usualmente altos, mientras que en el hipotiroidismo e hipercortisolismo se observa talla baja o deceleración del crecimiento.
Conclusión: Nuestro caso apoya la necesidad de evaluar detenidamente los signos físicos y el patrón de crecimiento en los niños obesos. En la mayoría de casos, los estudios deben limitarse a la analítica rutinaria de sangre, T4 libre y TSH. Una evaluación endocrinológica más completa debe reservarse para aquellos casos en que la observación clínica aporte datos relevantes, o si está disminuida la velocidad de crecimiento.
Obesidad en la adolescencia, riesgo cardiovascular. Valorar tratamiento con metformina
N. Cabrinety Pérez, M.ª J. Pisonero, A. Armenteras y J. Ajram
Servicio de Pediatría. Endocrinología Pediátrica. Hospital Sagrat Cor. Barcelona. España.
Introducción: La ob esidad se ha convertido en un grave problema de salud pública creciente, alcanzando niveles de pandemia en algunos países. La obesidad es un factor de riesgo importante tanto para la mortalidad prematura como para el desarrollo de importantes complicaciones médicas. En la adolescencia representa un importante riesgo de diabetes tipo 2 y de enfermedad cardiovascular. La metformina es una dimetilbiguadina que requiere la presencia de insulina para ejercer sus efectos sobre los tejidos periféricos insulinorresistentes, por lo que se ha postulado como un candidato como fármaco antiobesidad.
Objetivo: Valorar los efectos sobre el peso corporal (IMC) Hiperinsulinismo con o sin acantosis nigrigans y riesgo cardiovascular del tratamiento con metformina en un grupo de adolescentes obesos.
Pacientes y métodos: Se estudiaron 106 adolescentes obesos (59 niñas y 47 niños) con edad media de 12 a 14 años (13,1 ± 2,1) con IMC > P90 (32 ± 1,8). El estudio se realizó a lo largo de un año, se les determinó insulina basal, glucemia basal, HOMA, sobrecarga oral de glucosa, colesterol total y C-hdl, C-ldl, (presión arterial) PA d/s y función hepática. Se solicitó consentimiento paterno-tutelar informado. Se les distribuyó en tres grupos: 1.er grupo: 37 pacientes (21 niñas y 16 niños) a los que se les inició un programa de cambio del estilo de vida con dieta y ejercicio. 2.º grupo: 35 Pacientes (19 niñas y 16 niños) se les sometió al mismo programa recibiendo además metformina a dosis de 850 mg 2 veces al día. 3.º grupo: 34 pacientes (19 niñas y 15 niños) que sólo recibieron metformina en la misma dosis antes indicada.
Resultados: Al finalizar el estudio se observó una pérdida de peso en el 3.º grupo sometido sólo a tratamiento con metformina (media-14,9 kg ± 2,6) aunque estadísticamente no significativo a la perdida de peso observada en el 1.er Grupo tratado (media 16,1 kg ± 3,2) p < 0,05. La pérdida de peso más significativa fue del 2.º grupo tratado con metformina, dieta y cambios del estilo de vida (media 22,3 kg ± 2,5) siendo superior la reducción del consumo reducción del consumo calórico p < 0,0001. Los índices de riesgo cardiovascular y de sensibilidad a la insulina (HOMA, colesterol, etc.) mejoraron en los tres grupos, y fue significativamente mejor en el grupo 2, p < 0,0001, y no significativamente diferente en los otros dos grupos. El grupo 3 presentó una mejoría superior al grupo 1 con relación a la sensibilidad a la insulina no significativa p < 0,05. La TA d/s mejoró en los dos grupos sometidos a dieta p < 0,0001, pero no se observó cambio en los tratados sólo con metformina. De 62 pacientes que presentaban acantosis nigrigans (33 niñas y 29 niños) se redujo al mejorar los índices de sensibilidad a la insulina y en 12 pacientes (5 niñas y 7 niños) desapareció por completo. Los tres grupos toleraron bien el tratamiento sin ningún abandono y su función hepática no se alteró.
Conclusiones: El tratamiento de elección de la obesidad sigue siendo la dieta y los cambios en el estilo de vida. La utilización conjunta de metformina con dieta y los cambios en el estilo de vida parecen tener efectos beneficiosos sobre la pérdida de peso, reduciendo los lípidos, las concentraciones elevadas de insulina y otros factores de riesgo cardiovascular, sin ejercer efecto sobre la PA. La metformina podría estar indicada como tratamiento coadyuvante en pacientes con obesidad, hiperinsulinismo y riesgo cardiovascular.
Concentración de homocisteína, ácido fólico y vitamina B12 en el síndrome de Down
I. Echevarría Matía 1, J.I. Labarta 1, S. Conde 1, S. Martínez 2, J. Gracia 3, M.I. Salazar 4, A. Guallar 5, A. Baldellou 2, E. Mayayo 1 y A. Ferrández 1
1Unidad de Endocrinología. 2Unidad de Enfermedades Metabólicas. 3Servicio de Cirugía Pediátrica. 4Servicio de Bioquímica. 5Servicio de Medicina Nuclear. Hospital Universitario Miguel Servet. Zaragoza. España.
Introducción: La concentración de homocisteína se relaciona con el riesgo vascular y la enfermedad de Alzheimer, siendo ambas patologías más frecuentes en la población con síndrome de Down. En varios estudios se ha analizado la asociación entre las concentraciones de homocisteína y el ácido fólico, pero muy pocos estudios se han realizado en población infanto-juvenil.
Objetivo: Analizar la concentración plasmática de homocisteína, ácido fólico y vitamina B12 en individuos con síndrome de Down.
Material y métodos: Estudio descriptivo, prospectivo de 150 muestras procedentes de 79 pacientes con síndrome de Down de edades comprendidas entre 2 meses y 58 años controlados en nuestra unidad. La concentración de homocisteína plasmática (μmol/l) se realiza mediante ensayo de inmunofluorescencia polarizada y la concentración de ácido fólico (ng/ml) y vitamina B12 (pg/ml) se determina mediante inmunoanálisis electroquimioluminiscente. Se comparan las muestras con las de población control del Servicio de Bioquímica del hospital. Los cálculos estadísticos se realizan con el programa SPSS para Windows 9.0.
Resultados: En la tabla 1 se muestran las concentraciones de homocisteína, ácido fólico y vitamina B12 en la población infanto-juvenil frente a la población adulta.

En individuos con síndrome de Down las concentraciones de homocisteína plasmática aumentan significativamente con la edad (p < 0,001). En el grupo de edad adulta las concentraciones de ácido fólico y vitamina B12 son menores que en edades infantiles (p < 0,05).
La tabla 2 refleja las concentraciones de homocisteína, fólico y vitamina B12 por grupos de edad en la población infantil.
Si se comparan con población control las cifras de homocisteína en el grupo de edad de 10-14 años son significativamente más elevadas (p < 0,01).
El estudio descriptivo en el grupo de edad de 0-5 años demuestra los siguientes resultados:
a) En el 68 % de las muestras la homocisteína es mayor a 4,89 (IC 95 %: 4,12-4,89).
b) El 44 % presenta niveles de ácido fólico inferiores a 9,02 (IC 95 %: 9,02-12,46).
c) n el 64 % los niveles de vitamina B12 son inferiores a 864 (IC 95 %: 864-1.051).
Conclusiones:1) Nuestros resultados sugieren que en el síndrome de Down las cifras de homocisteína plasmática aumentan con la edad, al igual que sucede en la población control; pueden influir en sus concentraciones la cantidad plasmática de ácido fólico y vitamina B12. 2) En el grupo de edad de 10-14 años los niveles de homocisteína son significativamente más altos que en la población control. 3) Se necesitan estudios longitudinales a largo plazo para conocer el significado clínico de estos hallazgos.
Lipodistrofia, resistencia insulínica y acantosisnigricans
A. Fuentes Ortiz, M. Berrocal Castañeda, L. Prieto Tato, E. Álvarez, J. Cedeño Montaño y J. Prieto Veiga
Servicio de Pediatría. Departamento de Endocrinología. Hospital Universitario de Salamanca. España.
Introducción: La lipodistrofia es la pérdida y/o distribución anómala del tejido adiposo subcutáneo y/o visceral. Hay formas adquiridas de supuesto origen autoinmune. Las formas congénitas se heredan de forma AR y con menor frecuencia AD o ligada al cromosoma X. Se atribuyen a mutaciones de genes situados en las posiciones 9q34 y 11q13, que provocarían la apoptosis del adipocito. La disminución de la leptina podría tener un papel importante en la producción de las alteraciones metabólicas, siendo las más importantes las dislipemias y la resistencia insulínica.
Caso clínico: Niña de 10 años. Padres sanos no consanguíneos. Nació a término mediante cesárea (HTA materna y circulares de cordón) con peso, talla y PC adecuados a su EG. Peso y talla siguieron en P50-P75. A los 7 años comenzó a presentar un ensanchamiento progresivo de cara, cuello y hombros. Presenta apneas durante el sueño y mal rendimiento escolar (apoyo) con enuresis nocturna. Fenotipo peculiar: obesidad cushingoide facial, cervival y de cintura escapular y tórax, que contrasta con el aspecto grácil de las extremidades, en las que se observan glúteos, cuádriceps y gemelos prominentes. Llamativas lesiones de acantosis nigricans en cuello, axilas y periné. Estadio de Tanner: P1-T1 (adipomastia). Facies adenoidea. Hipertrofia amigdalar y de adenoides. Impresiona de cierto retraso psíquico. Exámenes neurológico y cardiológico normales. TA: 80/50. Antropometría: 134,2 cm (P37), 37.100 g (< P75), IMC: 20,7 kg/m 2 (P75). Exámenes complementarios: Hemograma, iones, perfil renal, albúmina, CK y osmolaridad plasmática normales. AST 48 U/l, ALT 81 U/l, LDH 539 U/l. Perfil lipídico: colesterol 225 mg/dl, HDL-C 30 mg/dl, LDL-C 151 mg/dl, TGs 189 mg/dl, índices de riesgo aterogénico altos. Perfil diabetológico: HbAC 4,8 %, fructosamina 183 umol/l, SOG: 100, 171, 126, 168 mg/dl. Insulinemia: 45, > 320 nmol/l. Autoinmunidad pancreática, tiroidea y degestiva negativas. TSH, prolactina, ACTH, cortisol basal, ritmo de cortisol y cortisol libre urinario normales. FSH y LH basales 6 y 0,1 mlU/ml. Leptina 9,8 ng/ml. IGF-1 115 ng/ml, IGF-BP3 4,1 μg/ml. Cariotipo 46 XX. Mapa óseo normal. EO 10,5 años (GP). Ecografía pélvica: ovarios grandes con patrón folicular normal. Polisomnografía: apneas frecuentes y prolongadas durante el sueño. Composición corporal (DEXA) en curso.
Discusión: la obesidad regional con escaso panículo distal y aparente hipertrofia muscular y el perfil psicológico sugieren una lipodistrofia congénita generalizada tipo Berardinelli Seip Trygstad (1954 y 1963), la cual se asocia habitualmente a síndrome metabólico con dislipemia, resistencia a la insulina y acantosis nigricans. En algunos casos se describen también concentraciones anormalmente bajas de leptina como consecuencia de la apoptosis de adipocitos. Aunque no hay signos clínicos ni ecográficos de adelanto puberal, el aumento de FSH sugiere un próximo crecimiento folicular. En cuanto al tratamiento, se ha optado por el empleo de metformina (20 mg/kg/día) junto con medidas higiénico-dietéticas con el fin de dificultar la evolución a ovario poliquístico, diabetes mellitus tipo 2 y esteatosis visceral. Es aconsejable la adenoidectamía y monitorización del sueño, en previsión de pausas de apnea graves. Estudios recientes han encontrado buenos resultados en pacientes que recibieron tratamiento con leptina con carácter experimental.
Evolución puberal de diferentes índices de insulinorresistencia en niños y adolescentes obesos
G. Bueno Lozano, L. Moreno Aznar, M. Viñas Viña, B. Tresaco Benedí, O. Bueno Lozano, J.M. Garagorri y M. Bueno Sánchez
Unidad de Endocrinología Pediátrica. Hospital Clínico Universitario. Zaragoza. España.
Objetivo: Establecer la prevalencia del síndrome de insulinorresistencia (obesidad, alteración hidrocarbonada, dislipidemia e hipertensión) en un grupo de 154 niños y adolescentes obesos. Estudiar el comportamiento de diferentes índices de insulinorresistencia en prepúberes (Tanner I) y púberes (Tanner II a V).
Pacientes y métodos: Se han estudiado 84 varones obesos (43 Tanner I, 30 Tanner II-III y 11 Tanner IV-V) y 70 mujeres obesas (20 Tanner I, 27 Tanner II-III y 23 Tanner IV-V), con una edad media de 10,83 ± 2,5 años. Como criterio de obesidad se han seguido las propuestas por Cole et al. Todos ellos fueron sometidos a una prueba de tolerancia oral de glucosa (1,75 mg de glucosa/kg), con determinación de glucemia e insulinemia basales y a los 120 min. Como variables del síndrome de insulinorresistencia se han incluido: perímetro de cintura, tensión arterial, glucosa, insulina, ácido úrico, triglicéridos y HDL-colesterol. La sensibilidad a la insulina a partir de determinaciones basales incluyó como índices: HOMA-IR (Homeostatic Model Assessment: insulinemia en ayunas [μU/ml] x glucemia en ayunas [mmol/l]/22,5), QUICKI (Quantitative Insulin Check Index: 1/log insulinemia en ayunas [μU/ml] + log glucemia en ayunas [mg/dl]) y el FIGR (fasting insulin to insulin ratio: insulinemia en ayunas [μU/ml]/glucemia en ayunas [mg/dl]). Cifras elevadas de HOMA-IR indicarían resistencia a la insulina. Cifras elevadas en el índice QUICKI indicarían sensibilidad a la insulina.
Resultados: 14 (9 %) pacientes (8 varones, 6 mujeres) mostraron tolerancia alterada a la glucosa, no habiéndose diagnosticado ningún caso de diabetes tipo 2. En ambos sexos, los índices HOMA IR (3,6 ± 2,39 en varones y 4,17 ± 3,16 en mujeres), QUICKI (0,33 ± 0,03 en varones y 0,32 ± 0,03 en mujeres) y FIGR (0,16 ± 0,10 en varones y 0,19 ± 0,1 en mujeres) indicaron cifras de insulinorresistencia y se correlacionaron significativamente (p < 0,05) con el índice de masa corporal, perímetro de cintura, triglicéridos, glucemia e insulinemia en ayunas y con la insulinemia a los 120 min. Tan sólo en el sexo masculino se evidenció correlación significativa con el HDL-colesterol. Considerados globalmente, la progresión en el estadio puberal se asoció con un incremento en las cifras de los índices HOMA y FIGR y con un descenso del QUICKI. Este último evidenció un descenso estadísticamente significativo (p < 0,05) entre los estadios I y IV tanto en varones como en mujeres.
Conclusiones: Los índices basados en determinaciones basales HOMA-IR, QUICKI y FGIR se correlacion an significativamente con los diferentes componentes del síndrome de insulinorresistencia. Sin embargo, es necesario establecer cuáles son sus diferentes puntos de corte según sexo y estadio puberal.
Insulinorresistencia en pacientes no obesos en edad pediátrica
Y. Alins Sahún, M. López Capapé, N. Dedieu, A. Carcavilla Urqui, M. Alonso Blanco y R. Barrio Castellanos
Servicio de Pediatría. 1Unidad de Diabetes y Endocrinología pediátrica. Hospital Universitario Ramón y Cajal. 2Universidad Alcalá de Henares. Madrid. España.
Introducción: La resistencia insulínica es un factor de riesgo de diabetes tipo 2 y enfermedad cardiovascular potencialmente tratable. Aunque se asocia frecuentemente con obesidad (el 40 % de 383 niños obesos seguidos en nuestra unidad presenta resistencia insulínica), puede detectarse en niños con normopeso.
Objetivo: Evaluar las características clínico-bioquímicas de nuestros pacientes insulinorresistentes con IMC < 2 DE, con factores de riesgo familiares o personales de síndrome metabólico (SM).
Metodología: Se analizan los datos de 8 pacientes no obesos con hallazgo de insulinorresistencia en estudio rutinario. Se recogieron los antecedentes familiares de primer y segundo grado de SM (obesidad, DM-2 o diabetes gestacional, HTA esencial, alteración lipídica y cardiopatía isquémica en menores de 50 años) y personales (peso y talla al nacimiento, somatometría, estadio puberal). En todos los casos se realizó SOG, HbA1c, lipidograma, perfil hepático y, en el 75 %, ecografía hepática para descartar esteatosis. La sensibilidad y resistencia insulínicas durante la SOG se calcularon mediante el índice glucosa/insulina (Glu/Ins), el QUICKI (1/[log (insulina b) + log (glucosa b)], el HOMA-IR (Glu x Ins/22,5) y el ISI (1/Insulina 120 min). La TA se evaluó con las tablas de la Task Force (2004). Se utilizaron los criterios de Cook (2003) modificados para la valoración de síndrome metabólico en adolescentes. La alteración hidrocarbonada se valoró según consenso de la ADA 2004 (glucemia basal > 100 y 2 h > 140 mg/dl). La tabla 1 resume las características clínicas y analíticas de estos pacientes.
Resultados: La edad media de los pacientes fue de 11,66 ± 2 años (el 87,5 % mujeres, 6 en edad puberal), ninguno PEG. Todos los pacientes tenían antecedentes familiares de SM. En 6 pacientes se detectaron criterios de síndrome metabólico: 4 casos con dos criterios (50 %) y 2 con tres criterios (25 %). El 62,5 % del total presentaba alteración hidrocarbonada, y en el 50 % de las ecografías hepáticas realizadas (3/6) se objetivaron signos de esteatosis (en el caso número 6, con aumento de GOT y GPT). En todos los pacientes se intensificó el control de los factores de riesgo (peso, actividad física, etc.). El paciente número 7 recibió tratamiento con metformina durante 2 años, normalizándose la SOG.
Conclusiones:1) Los antecedentes familiares de síndrome metabólico deben ser cuidadosamente evaluados en todos los pacientes, ya que constituyen un marcador de riesgo para la insulinorresistencia en la edad pediátrica. 2) En el momento del diagnóstico de la resistencia insulínica están presentes otros componentes del síndrome metabólico. 3) La alteración hidrocarbonada en estos pacientes puede aparecer precozmente. 4) En casos seleccionados el tratamiento con metformina oral puede revertir la insulinorresistencia.
Distribución regional de la grasa corporal en bailarinas de ballet clásico. Relación con los niveles de las proteínas leptina y ghrelin
M.A. Donoso Sanz, M.T. Muñoz-Calvo, V. Barrios, G. Garrido, G. Martínez, F. Hawkins y J. Argente
Servicio de Endocrinología. Universidad Autónoma. Hospital Infantil Universitario Niño Jesús. Madrid. España.
Introducción: El tejido adiposo desempeña una función relevante en el equilibrio energético del organismo, merced a la producción de diferentes factores hormonales Entre éstos, destaca la leptina, que inhibe el metabolismo y el apetito. La proteína ghrelin, producida fundamentalmente en el tracto gastrointestinal, tiene acciones contrapuestas, estimulando el metabolismo y el apetito.
Objetivos:1. Analizar los cambios en la composición corporal (masa muscular, grasa corporal total y distribución regional de la grasa) en un grupo bailarinas al inicio de la pubertad. 2. Relación con los niveles de leptina y ghrelin.
Sujetos y métodos: Se estudiaron 25 bailarinas de ballet clásico (B), con una edad media de 11 ± 1 años, en estadio II de Tanner, que realizaban 12 h de ejercicio físico a la semana. El grupo control (C) estaba formado por 10 niñas de similar estadio puberal y edad. A todas se les realizó un estudio antropométrico: peso (kg), talla (cm), índice de masa corporal IMC (expresado en DE para la edad y el sexo), y se determinó la edad ósea (Greulich y Pyle). Así mismo, se realizó una encuesta nutricional de 5 días (Nutrionist®) y se cuantificó la masa muscular (g), y el porcentaje de grasa corporal total (% GCT) y grasa regional [grasa de tronco (% GT) y de extremidades (% GE)] por método DEXA (Hologic 4500). Leptina y ghrelin se determinaron por RIA (Linco, EE.UU.).
Resultados: El aporte energético fue similar en los dos grupos (RDA/RDI), a pesar de la diferente actividad física. En el grupo de bailarinas, la masa muscular fue normal. Por el contrario, los porcentajes de GCT, GT y GE fueron significativamente más bajos con respecto al grupo control. Los niveles de leptina estaban significativamente disminuidos (p < 0,01), mientras que los de ghrelin fueron normales. El IMC y la edad ósea se encontraron dentro de los límites de la normalidad. Se observó una correlación significativa entre los niveles de leptina con el IMC, porcentajes de GCT, GT, y GE (r = 0,78, r = 0,46, r = 0,57, r = 0,46, respectivamente; p < 0,01). No se encontró correlación de la grasa corporal con los niveles de ghrelin (tabla 1).

Conclusiones: 1) El ejercicio físico intenso, a pesar de un aporte energético relativo al peso equilibrado, produce una pérdida de la grasa corporal total y una disminución proporcional de la grasa regional. 2) La leptina es un buen marcador periférico del porcentaje de grasa corporal.
Hipoglucemia hiperinsulínica por nueva mutación del genSUR1
M.ª J. García Arias, M.M. Serrano, M. Laakso y J.P. López-Siguero
Servicio de Endocrinología Pediátrica. Hospital Materno-Infantil. Hospital Universitario Carlos Caya. Málaga. España. Universidad de Kuopio. Finlandia.
Motivo de consulta: Hipoglucemias desde las 2 h de vida que no se resuelven con aportes orales de glucosa. Antecedentes familiares: madre con diabetes gestacional. Antecedentes personales: embarazo de 36 semanas con bajo peso para EG: 2.180. Apgar 9/10. Exploración inicial: PRN: 2.180 g. LRN: 45 cm. No hay datos patológicos. Evolución y datos complementarios: hipoglucemias detectadas por pauta de control glucemico (hija de madre diabética). Nunca presentó clínica. Las hipoglucemias persisten y precisan para su control aportes de glucosa IV de hasta 22 mg/kg/minuto, además de alimentación continua (sonda nasogástrica) con leche materna. Las hipoglucemias coinciden con niveles de β -OH-butirato de 0 a 0,1 mmol/l, con ácidos grasos libres de 0,29 mE/l (0,3-0,5). Se realizó un estímulo con glucagón en hipoglucemia, y se obtuvo un incremento en la glucemia de 55 mg. Amoniemia: 51 μmol/l (20-57).

La paciente fue tratada con diazóxido (sin respuesta), octeótrido subcutáneo en dosis de 10 μg/kg/dosis, con respuesta positiva, aunque parcial porque presentaba hipoglucemias ocasionales, que fueron tratadas con glucosa IV o glucagón. Mantuvo una alimentación materna continua suplementada con dextrinomaltosa a través de sonda NG. A partir de los 2 meses de vida se observa una desaparición de las hipoglucemias que permiten una lenta y progresiva retirada del tratamiento. El desarrollo psicomotor, EEG y ecografia craneal han sido normales. Análisis molecular: Se encontró una delección en el codon 1192 (GAT) en el gen SUR1 (1192delG), confirmada por secuenciación. Esta alteración no está descrita hasta la fecha y produce una alteración grave de la secuencia proteica.
Comentarios: Dada la evolución de la paciente y el resultado del estudio genético, el diagnóstico concuerda con un hiperinsulinismo persistente en forma focal. Se especula que la desaparición (por apoptosis) de las células b afectadas sería la responsable de la normalización glucémica, pudiendo producir en el futuro una diabetes insulinopénica.
SUPRARRENALES Y OTRAS
Estudio descriptivo de las características clínicas y hormonales de la adrenarquia precoz en el momento del diagnóstico. Prevalencia de formas no clásicas de déficit de 21-hidroxilasa
E. Vera de Pedro, I. Díez López, B. Orive Olandriz, E. Gracia Ojeda, G. Martínez de la Hidalga, I. Ocio Ocio y A. Rodríguez Estebez
Servicio de Pediatría. Hospital de Txagorritxu. Álava. España.
Objetivo: Estudio descriptivo de las características clínicas y de los valores de andrógenos basales y a los 60 min del estímulo con ACTH (0,25 mg IV) de los niños diagnosticados en nuestro centro de adrenarquia precoz entre los años 1989 y 2005. Prevalencia de formas no clásicas de déficit de 21-hidroxilasa (21-OH) en esta población.
Material y métodos: 140 niños (23 V y 117 M) con adrenarquia precoz (aparición de vello pubiano antes de los 8 años en niñas y 9 años en niños). Al diagnóstico se valoraron aspectos clínicos (axilarquia, telarquia y volumen testicular), auxológicos (edad ósea) y se realizó estudio de andrógenos basales (17-hidroxiprogesterona [17 OHP] y el 11-desoxicortisol [11 Desox], DHEA, DHEA-S, Delta 4-androstendiona [D4-A] y Testosterona [T]) en 42 niños (30 %) y prueba de estimulación con ACTH en 98 niños (70 %) con 17 OHP y 11 Desox basal y postestímulo y el resto de andrógenos basales. Diagnosticados de déficit no clásico de 21-OH si 17-OHP postestímulo superior a 15 ng/ml (15-100) y heterocigotos entre 3 y 14,9 ng/ml. En todos los niños con 17-OHP basal > 2 ng/ml se realizó el test de ACTH. Dada la importante superposición que se produce entre no afectados y heterocigotos, hemos estudiado únicamente aquellos heterocigotos con valores de 17-OHP postestímulo superiores a 10 ng/ml.
Resultados: Presentaban una edad media al diagnóstico de 7 años y edad ósea (método de Greulich-Pyle) al diagnóstico de 8,6 ± 2,0DE. La edad ósea se encontraba adelantada 1,2 años (± 2,0 DE) con respecto a la edad cronológica. Asociaban axilarquia el 27 % de los pacientes y telarquia el 8 % de las niñas; ningún varón había iniciado pubertad. Los valores del estudio hormonal se muestran en la tabla 1.
En este grupo de adrenarquia precoz se diagnosticaron 3 niños (3 V) de déficit no clásico de 21-hidroxilasa (2,15 %) casos 4, 5, y 6, y 3 niñas (3 M) (2,15 %) de heterocigotas con 17-OHP postestímulo superior a 10 ng/ml, aunque fueron 29 niños los que presentaron un valor de 17-OHP post-ACTH ≥ 3 ng/ml (29,6 % de heterocigotos del total de test de ACTH realizados). En el grupo de formas no clásicas y heterocigotos seguros, excepto en el caso 3, existían antecedentes familiares de hiperandrogenismo en padres y hermanos, predominantemente hirsutismo y acné. En el caso 4 la madre tenía un déficit no clásico de 21-hidroxilasa. En todos la edad ósea se hallaba incrementada más de 2 años (media 2,76 años) excepto en el caso 4 que fue diagnosticado a la edad de 2 años y presentaba un incremento de 1 año en la maduración ósea. En el caso 1 con el mayor avance en la maduración ósea se asociaba en el momento del diagnóstico una pubertad precoz, existiendo además este antecedente en una hermana. Los datos de estos niños se reflejan en la tabla 2.
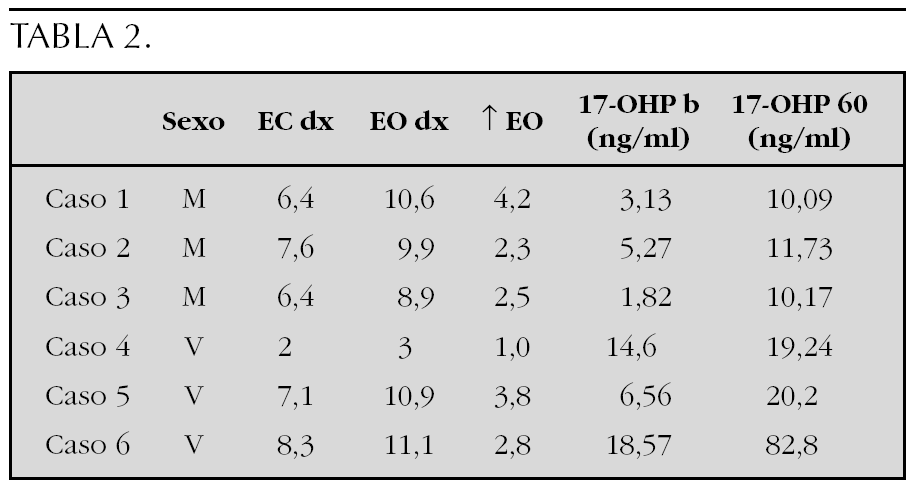
Conclusiones:1) Prevalencia del 2,15 % de formas no clásicas de déficit de 21 hidroxilasa en nuestros niños con adrenarquia precoz. 2) Todos los niños con déficit no clásico de 21-OH fueron varones (13 % del total de varones de la serie). 3) 2,15 % fueron heterocigotos con criterio estricto de > 10 ng/ml post-ACTH, todas mujeres (2,5 % del total de mujeres), si bien con criterio habitual un 29,6 % serían heterocigotos en nuestra serie. 4) Incremento mayor de la edad ósea en este grupo (2,76 frente a 1,2) de formas no clásicas-heterocigotos seguros de 21-hidroxilasa.
Correlación entre fenotipo y genotipo en hiperplasia suprarrenal congénita. Predictores clínicos de mutación severa
P. Cabanas Rodríguez 1, J. Barreiro 1, L. Loidi 2, L. Castro-Feijoó 1, T. Arévalo 1, M. López-Franco 1, F. Vasconcelos-Espada 1, C. Quinteiro 2, F. Domínguez 2 y M. Pombo 1
1Unidad de Endocrinología. Crecimiento y Adolescencia. Hospital Clínico Universitario de Santiago. A Coruña. 2Unidad de Medicina Molecular. FPGMX. España.
Introducción: La hiperplasia suprarrenal congénita (HSC) es un trastorno de la biosíntesis adrenal de colesterol, siendo su causa más frecuente la deficiencia de actividad de la enzima 21-hidroxilasa (21-OH). Las delecciones y otras alteraciones cromosómicas en el gen CYP21 han sido identificadas como causa de déficit de 21-OH. Estas mutaciones han sido clasificadas según el grado de déficit enzimático.
Objetivos: Describir predictores clínicos de presencia o ausencia de mutaciones severas.
Material y métodos: Se estudiaron los pacientes diagnosticados de HSC por déficit de 21-OH en nuestro centro, clasificándolos en dos grupos según presencia o ausencia de alguna mutación severa determinada por análisis genético molecular. Una vez establecidos mediante análisis de regresión logística los mejores predictores clínicos y bioquímicos de mutación severa, se establecieron a partir de la curva ROC asociada los puntos de corte óptimos para el diagnóstico de presencia o ausencia de mutación severa.
Resultados: Se incluyeron 55 pacientes (11 varones), 22 de ellos con presencia de al menos una mutación severa.
Los mejores predictores fueron: edad ósea, talla diana, clitoromegalia o macrogenitosomía, 17-OH-progesterona, androstendiona y cortisol basales. Determinamos la función de regresión logística que describe la correlación entre estas variables para identificar pacientes con o sin mutación severa. La curva ROC derivada de esta ecuación tiene un área bajo la curva de 0,95 (p = 0,003) con dos puntos de corte óptimos, el primero con sensibilidad 100 % y valor predictivo positivo 71,43 %, y el segundo con especificidad 100 % y valor predictivo negativo 88,89 % para la identificación de pacientes con mutaciones severas.
Conclusiones: El fenotipo clínico y bioquímico de los pacientes con HSC por déficit de 21-OH permite predecir la presencia de mutaciones severas.
Hipoplasia adrenal congénita e hipogonadismo hipogonadotropo por una nueva mutación del genDAX-1en el codón 184 (g 2080-2081 INS a)
J.L. Ruibal Francisco, O. Pérez Rodríguez, L. Loidi Fernández de Trocóniz y S. Parajes Castro
1Servicio de Pediatría. Hospital Clínico Universitario San Carlos. 2Medicina Molecular. Hospital Clínico de Santiago de Compostela. España.
Introducción: La hipoplasia adrenal congénita agrupa varias enfermedades que tienen en común el incorrecto desarrollo de las glándulas suprarrenales durante el período embrionario. En el momento actual se conoce la existencia de varios genes que intervienen en la morfogénesis de la corteza suprarrenal entre los que se encuentra el gen DAX-1, que se localiza en Xp21. Codifica una proteína que pertenece al grupo de receptores nucleares huérfanos y se denomina NORB1. Es un factor de transcripción de 470 aminoácidos y se expresa en las glándulas adrenales, en las gónadas y en la región hipotálamo-hipofisaria. Presentamos el caso de un varón de 18 años con una hipoplasia adrenal y un hipogonadismo hipogonadotropo por una mutación del gen DAX-1 no descrita hasta el momento.
Paciente estudiado: Joven de 18 años de edad sin antecedentes familiares de interés, cuya historia comienza en el período neonatal. Fue producto de un embarazo gemelar monocorial y monoanniótico, cuyo hermano gemelo pensamos que falleció de una insuficiencia suprarrenal en el seno de la misma enfermedad que padece este paciente. Ingresó a los 16 días de vida por presentar un cuadro de insuficiencsíndrome pierde-sal (Na = 110 mEq/l) con hiperpotasemia (K = 7,5 mEq/l). Fue tratado con líquidos y electrolitos, glucocorticoides y aporte continuo de sal. Se mantuvo sin tratamiento con un estado de salud aceptable hasta los 2 años de edad, en que comenzó con una sintomatología progresiva consistente en anorexia, avidez por la sal, desmedro, hiperpigmentación cutánea y finalmente un cuadro de choque, hipoglucemia, hiponatremia grave, acidosis metabólica e hiperpotasemia, siendo diagnosticado de insuficiencia suprarrenal global que se trató con y líquidos y electrolitos y aportes de gluco y mineralcorticoides de manera permanente. Para dilucidar la etiología del insuficiencia suprarrenal se procedió a los 11 años de edad a la retirada progresiva de los mineral y glucocorticoides. En situación de insuficiencia suprarrenal se procedió a la extracción de sangre para valoración de la función suprarrenal.
Resultados: ACTH 1.450 pg/ml (N = 9-52); 17-OH-progesterona, cortisol, D-4-androstendiona basales y tras 250 μg de ACTH sintética IV indetectables; renina 1900 μUI/ml y aldosterona indetectable. A los 13,4 años se realizó un test de LHRH que mostró una LH y FSH basales indetectables y de 2,2 y 3,8 mUI/ml, respectivamente, tras estímulo. Además de la terapia sustitutiva suprarrenal, se inició tratamiento con preparados depot de testosterona. Su talla final ha sido de 150 cm (talla diana 164 cm), no presentó desarrollo puberal espontáneo siendo el volumen testicular definitivo alcanzado de 2 ml y una pubarquia de V. El estudio genético en sangre con amplificación PCR para el gen DAX-1 mostró una mutación g 2080-2081 ins a en la primera posición del codón 168, produciendo una parada prematura de la proteína DAX en la posición 184.-
Conclusiones: Se presenta una nueva mutación del gen DAX-1 en un paciente que presentó insuficiencia suprarrenal completa en el período neonatal, ausencia de pubertad y talla baja patológica. Las nuevas mutaciones del gen DAX-1 ayudan a comprender la relación fenotipo-genotipo en cuadros de hipoplasia adrenal congénita.1
Relación genotipo-fenotipo en HSC
I. Leiva Gea, A. Caro Jaén, E. Caro Cruz, E. García-Triviño Arboleda, J. de la Cruz Moreno, M.D. Gámez Gómez, A. Leiva Gea y A. Arregui Armenteriz
Servicio de Pediatría. Complejo Hospitalario de Jaén. España.
Introdución: La hiperplasia suprarrenal congénita (HSC) en el 90 % de los casos se debe a déficit de 21-hidroxilasa con una prevalencia de 1/12.000 en la forma clásica y de 1/100 a 1/1.000 en la forma no clásica. Es una enfermedad hereditaria monogéncia AR. Según su forma de presentación clínica se clasifican en: perdedora de sal (SW), virilizante simple (VS) o forma tardía, también denominada no clásica (NC). La frecuencia de portadores es de 1/50 en formas severas y de 1/5 en formas tardías, sin olvidar que las formas tardías pueden ser portadoras de mutaciones severas. En la forma clásica el aumento de 17-OH progesterona es generalmente 10 veces el valor normal, siendo en las formas no clásicas la elevación más leve. Cifras menores de 2 ng/dl descartan el diagnóstico, y éste es probable con cifras mayores de 6 ng/dl. Se recomienda test de ACTH cuando los valores de 17-OH progesterona se encuentran entre 2 y 6 ng/dl, considerando como anormal una respuesta superior a 10 ng/dl. La prevalencia de formas no clásicas de hiperplasia suprarrenal congénita en niños con pubertad precoz es 7-10 %. Las formas no clásicas de la deficiencia constituyen la patología herediataria más frecuente en nuestro medio. En el estudio genético de esta entidad, las mutaciones más frecuentemente observadas son: intrón 2 en el 50 % de la forma perdedora de sal (SW) y en el 50 % de forma virilizante simple (VS), I 172 N en el 16,6 % de los casos de forma virilizante simple y V 281 L en 61,5 % de casos de formas no clásica (NC).
Material y métodos: Se revisan 13 casos de HSC en nuestro medio. Se presentan 5 SW, 1 VS y 7 NC. Los pacientes fueron clasificados de acuerdo al cuadro clínico y el diagnóstico confirmado por dosis hormonales. Se realizó estudio molecular a 11 de los 13 casos junto con sus familias correspondientes. En los 3 casos de forma WS en los que se realizó estudio genético, las alteraciones encontradas fueron: intrón 2G/Mutación no detectada, intrón 2/intrón 2 y delección-conversión intrón 2/Exón 3/Del 8 pb. En el caso con fenotipo VS el genotipo observado es intrón 2 G/Normal.
Resultados: En los casos con fenotipo NC se realiza estudio genético a los 7 casos, con los siguientes resultados: Val 281 l/ Val 281 l (2/7), Glu 318 Stop/Normal (1/7), Val 281 l/normal (3/7) y Val 281 l/mutación no detectada (1/7). En el estudio genético y hormonal de los familiares se detectan 8 formas crípticas. Se entiende por formas crípticas, sujetos asintomáticos con leve hiperandrogenismo biológico y todos ellos portadores de anomalías en el gen de la 21-OH en uno o los dos cromosomas; 7 de las 8 formas crípticas presentan alteraciones tan sólo en uno de los cromosomas, siendo la mutación más frecuentemente encontrada Val 281 l/normal; 1 hermano de la forma WS se muestra portador de una mutación severa (delección-conversión intrón 2/exón 3/normal). Mostrando afectación en sus dos cromosomas sólo un caso, hermano de forma no clásica, con genotipo (Val 281 l/Val 281 l).
Conclusiones:1) Del análisis de la relación genotipo/fenotipo se observa una alta correlación entre las manifestaciones clínicas y el genotipo encontrado. La mutación más frecuentemente observada está en la forma SW intrón 2 al igual que en la forma VS y Val 281 L en forma no clásica. Se corresponde con los resultados encontrados en otras series estudiadas. 2) Importancia del estudio de familiares de los casos afectados que permite el diagnóstico de formas crípticas de la deficiencia que son susceptibles de consejo genético. 3) El hecho de observar dentro de una misma familia hermanos afectados con idéntico genotipo CYP21 pero con diferente forma clínica (forma NC y forma críptica) hace pensar que otros factores genéticos o ambientales deben estar implicados en la variabilidad de esta expresión clínica, apoyada por las observaciones de otros estudios que apoyan esta interpretación.
Composición corporal y resistencia periférica a la insulina. Su relación con el hiperandrogenismo de la HSC por déficit de 21-hidroxilasa
A. Oliver Iguácel, C. Oliver Vigueras, R. Lama More, B. Lodoso Tordesillas, J. Guerrero Fernández y R. Gracia Bouthelier
Servicio de Endocrinología Pediátrica. Hospital Infantil La Paz. Madrid. España.
Introducción: La terapéutica de supresión con cortisona en la HSC es limitada y en general, se mantienen niveles elevados de andrógenos. Existe el riesgo de resistencia periférica a la insulina y alteraciones en la composición corporal secundarios al hiperandrogenismo de estos pacientes.
Objetivos: Conocer las características de composición corporal y su relación con las alteraciones metabólicas y hormonales en niños diagnosticados HSC por déficit de 21 hidroxilasa.
Material y métodos: Se estudian 68 niños: 32 controles con edad media de 10,5 años (7-17) sin alteraciones hormonales (17 niñas y 15 niños) y 38 pacientes (18 mujeres y 20 varones) diagnosticados de HSC por déficit de 21-hidroxilasa, 24 pacientes con forma no clásica y 14 con forma clásica. Edad media: 11,2 años (2-22). Se analiza la composición corporal por antropometría (fórmulas de Durning y Siri) y por Bioimpedancia (software RJL System). La insulina basal mU/ml, el HOMA (glucosa basal mmol/l x insulina basal μU/ml)/22,5 y la testosterona en pg/ml.
Resultados: Los niveles de Testosterona fueron muy variables: 1.766 ± 2.555 pg/ml, no objetivándose diferencias por la edad ni por el sexo de los pacientes. Insulina basal (IB) 15 ± 15 mU/ml y los de HOMA: 2,3 ± 1,77, habiéndose encontrado diferencias significativas entre púberes y no púberes (IB: p = 0,01 y HOMA p = 0,02) pero no encontramos diferencias por sexo. Aunque los niveles de insulina basal IB en pacientes fue mayor que en los controles (11,2 ± 4,5) esta diferencia no fue significativa. Los pacientes presentaron: los índices de relación peso/talla significativamente más altos que los controles, observamos, así mismo, que la masa corporal grasa (MCG) fue superior (105,7 ± 34 %N frente a 178 ± 73 % N p = 0,008), no encontrándose relación con la masa corporal magra MCM (27,3 ± 10 frente a 32,96 ± 14 p = 0,001). Había una correlación significativa entre los niveles de T y la MCM (p < 0,02 r = 0,4) pero no con la MCG. La MCG se correlacionaron con los niveles de IB (p = 0,009 r = 0,74) y de HOMA (p = 0,01 r = 0,68). Los niveles de T no se correlacionaron con con los de IB ni con HOMA.
Conclusiones:1) En nuestra serie se encontró un aumento de masa corporal grasa y ligero de MCM. 2) La MCG se correlacionó con la IB y con HOMA. 3) La MCM se correlacionó con la T. 4) No encontramos correlación entre los niveles de T y de IB ni HOMA 5) En nuestra serie, la resistencia periférica a la insulina no sería secundaria directamente al hiperandrogenismo.
Síndrome de Antley-Bixler e hiperplasia suprarrenal
J.A. Nieto Cuartero, S. Vila, A. Linares, A. Mínguez, M. Monleón y A. Pascual
Servicios de Endocrinología, Neurocirugía, Rehabilitación, Traumatología y Laboratorio RIA-UCIP. Hospital Infantil Universitario Niño Jesús. Madrid. España.
Introducción: Antley y Bixler describieron en 1975 la asociación de craneosinóstosis, sinóstosis radiohumeral y atresia coanal. En una reciente revisión se han descrito 23 casos en el mundo y el nuestro aporta la evolución tras tres años y medio de la asociación con hiperplasia renal por déficit de 21 hidroxilasa, hipospadias micropene criptorquidia bilateral, hipoacuasia neurosensorial y higroma cerebral, con inteligencia normal y colocación de válvula de derivación en el ventrículo cerebral peritoneal y la normalización con el terapia sustitutiva gluco y mineral corticoide de las cifras de testosterona sérica SDHEA 17-alfa-OH progesterona, aldosterona y renina. Antecedentes familiares: hijo único de padres no consanguíneos, sanos. Antecedentes personales: 35 semanas de gestación, parto por cesárea, PRN: 2,430 kg TRC: 48 cm. Las cifras de 17-alfa-OH progesterona estaban muy elevadas: 120 nmol/l y fueron subiendo a los 5 días a 193 nmol/l, se instauró tratamiento sustitutivo corticoideo, con lo cual se logró normalizar las cifras elevadas de 17-alfa-OHP, al igual que las cifras de ACTH, aldosterona, renina y SDHEA. El escáner cerebral mostró una platibasis severa con marcado aumento del espacio subaracnoideo y del volumen del sistema ventricular comunicante. Se colocó por el Servicio de Neurocirugía una válvula de derivación ventrículo peritoneal; igualmente, se realizó la corrección de la sinostosis radiohumeral cubital bilateral, por el Servicio de Traumatología. Se aprecia una campo dactilia de los dedos de la mano derecha, tercero, cuarto y quinto, caderas y rodillas en flexión pero reductibles.
Conclusiones:1) Aportamos la asociación de hiperplasia renal congénita, con micropene y criptorquidia al síndrome de Antley-Bixler. 2) Comentamos la excelente respuesta tanto a la corrección de la craneosinóstosis como del control de su hiperplasia suprarrenal y que la colaboración de neurocirujanos, traumatólogos, endocrinos pediátricos y rehabilitadores, logra una mejoría en la evolución de este síndrome.
Síndrome de Von Hippel Lindau
C. Rodríguez Delhi, M.ª J. Antuña, M. Galbe y M.F. Rivas Crespo
Endocrinología Pediátrica y Oncología Pediátrica. Hospital Universitario Central de Asturias. Oviedo. España.
Introducción: El feocromocitoma es un tumor endocrino infrecuente. Puede formar parte de síndromes familiares neoplásicos: la neoplasia endocrina múltiple 2 y el síndrome de von Hippel Lindau, ambos autosómicos dominantes.
Caso clínico: Niño de 12 años que consulta por pérdida de visión en el ojo izquierdo, refiriendo además poliuria, polidipsia, y sudoración. Su tensión arterial está elevada (198/135 mmHg) y en el fondo de ojo se objetiva una retinopatía hipertensiva de grado IV. En el estudio etiológico de la hipertensión, la función renal y hepática son normales, pero las catecolaminas urinarias están patológicamente elevadas y se ve una masa suprarrenal izquierda en la ecografía y la TC. Ésta es hipercaptante en la gammagrafía con 123I, identificándose como feocromocitoma, por lo que controlada la hipertensión arterial con fenoxibenzamina, se hace suprarrenalectomía izquierda. Tras ella, se mantiene normotenso sin precisar tratamiento. El estudio genético objetiva la mutación TC (175) AAC/Y175N del gen VHL (3p25-p26) en el paciente y en su madre. Tres meses después vuelven a elevarse las catecolaminas y la normetanefrina en orina; se descubre otro feocromocitoma en la glándula contralateral, que obliga a su extirpación. Actualmente está con tratamiento sustitutivo con hidraltesona, con controles periódicos por la posibilidad de nuevas neoplasias en los que se le ha detectado un angioma cerebeloso.
Comentarios: Es necesario realizar un estudio genético a todos los casos de feocromocitoma, especialmente si es bilateral, por la posibilidad de un síndrome familiar (asociado o no a otras neoplasias) por el riesgo de otras neoplasias asociadas en el propio paciente y en sus familiares. También es fundamental para su consejo genético.
Feocromocitoma: un tumor excepcional en niños
C. Del Castillo Villaescus, J. Marín Serra, J.L. Tortajada Soriano, M.J. López García, R. Alpera Lacruz y R. Hernández Marco
Servicio de Pediatría. Hospital Doctor Peset. Valencia. España.
Introducción: El feocromocitoma es un tumor muy poco frecuente. Aparece en 0,5-0,1 % de la población hipertensa y solamente un 10 % en niños, con predominio en varones. Se localiza en la médula suprarrenal en un 85 % de los casos, y es bilateral en el 20-25 %. La clínica viene dada por el aumento de noradrenalina o adrenalina procedente del tumor (HTA, cefalea y aumento de sudoración). Existe asociación familiar en el 10-30 % de los casos y raramente es maligno (menos del 10 %).
Caso clínico: Escolar mujer de 7 años remitida por cefalea de tres meses de evolución y alteración de la tensión arterial (sistólica de 130 mmHg). Pérdida de apetito y aumento de la sudoración en los últimos meses. Antecedentes familiares: Padre 36 años, hipercolesterolemia. Madre y hermana de 12 años sanas. Abuela materna, HTA, fallecida a los 67 años por hemorragia cerebral. Tío materno 40 años, HTA y manchas cutáneas. No antecedentes de tumores sólidos ni neurofibromatosis. Antecedentes personales: sin interés. Exploración física: Peso: 22,5 kg (P25). Talla: 128 cm (P90). T.ª axilar: 36 °C. FC: 100 l/m. PA (ambos miembros superiores): 175/124. Buen aspecto general, ligera palidez cutánea, pulsos periféricos simétricos. Auscultación cardíaca: soplo sistólico I/VI en borde esternal izquierdo y ápex, segundo ruido desdoblado fijo. Auscultación pulmonar normal. Abdomen sin masas ni hepatoesplenomegalia. No soplos en abdomen ni en fosa lumbar. Estudio neurológico normal. Genitales normales. Aumento de pigmentación en codos y ano. Seis manchas café con leche desde el nacimiento, una mayor de 2 cm. Exploraciones complementarias: Hemograma: serie roja normal, leucocitosis 16400 con neutrofilia 79 %. Bioquímica: glucemia 100 mg/dl, creatinina 0,4 mg/dl, GOT/GPT 29/25UI/l, colesterol total 268 mg/dl, calcio 10,4 mg/dl, fósforo 4,6 mg/dl, magnesio 2,7 mg/dl, fosfatasas alcalinas 162 UI/l, proteínas totales 8 g/dl, iones normales. TSH y T4L normales, cortisol 8h: 50 μg/dl, PTHi 33 pg/ml. Enolasa neuro-específica: 14,6ng/ml. Orina 24 h: ClCr 121 ml/min/1,73 m 2, calcio 6,7 mg/kg/día, Ca/Cr 0,33, EFNa 0,11 %, aldosterona 24 μg/24h, cortisol 177 μg/24 h, adrenalina 82 μg/día, noradrenalina > 7.500 μg/día, ácido vanilmandélico 14 mg/día. Ecografía abdominal/Doppler renal: masa suprarrenal derecha de 3,8 x 3,2 x 3 cm. Suprarrenal izquierda y riñones normales. RM: tumoración redondeada comprometiendo suprarrenal derecha, de 4 x 3 cm. En T2, tumor de alta intensidad, típico tumor cromafín. No otras lesiones. TC torácica: normal. ECG: normal; Ecocardiografía-Doppler: insuficiencia mitral leve con válvula normal. Gammagrafía ósea: normal. Gammagrafía con MIBG (10 h y 24 h tras trazador): no captación. Evolución: Con diagnóstico de sospecha de feocromocitoma se inicia tratamiento con nifedipina sin respuesta, por lo que se cambia a fenoxibenzamina; se trata durante 13 días con respuesta lenta, disminuyendo las cifras de PA de 150/87 a 130/80. Se realiza intervención quirúrgica el día 16 de tratamiento sin incidencias. Tras la intervención se reinicia tratamiento antihipertensivo (fenoxibenzamina) por cifras tensionales de nuevo elevadas. Disminuye la excreción de normetanefrina en orina de 24 h a 297 μg/día. El informe anatomopatológico confirma el diagnóstico de feocromocitoma adrenal.
Comentario: El feocromocitoma es una causa tratable de HTA que debe ser tenida en cuenta ante un niño con HTA, sobre todo si es mantenida. Tras el diagnóstico es importante el seguimiento del paciente y también el estudio familiar debido a la posible asociación con otros tumores, como los del grupo MEN y displasias neuroectodérmicas.
Disgenesia gonadal parcial
A. Domínguez García, S. Quinteiro González, N. Montesdeoca Araujo y Y. Novoa Medina
Servicio de Pediatría. Hospital Universitario Materno-Infantil de Las Palmas. España.
Introducción: La disgenesia gonadal mixta (DGM) y el seudohermafroditismo masculino disgenético (SMD) son dos formas de seudohermafroditismo masculino que se presentan con ausencia de regresión de estructuras mullerianas y genitales ambiguos. La DGM se caracteriza por gónadas, asimétricas que consisten en un testículo con grados variables de disgenesia en un lado y una cintilla gonadal, indiferenciada, en el otro, el cariotipo suele ser 45 X/46 XY aunque puede ser 46 XY. El SMD 46 XY se caracteriza por que ambas gónadas presentan diferenciación de tipo testicular, aunque sea disgenética.
Caso clínico 1: Niña de 10 años que consulta por estancamiento estatural en los últimos 3 años. Antecedentes personales (AP): sin interés. Antecedentes familiares (AF): talla materna 170 cm, talla paterna 175 cm, talla diana 166 cm (SDS 0,63). Exploración física: peso 39,2 kg, talla 128,5 cm (SDS 1,64), velocidad de crecimiento 1,1 cm/año (p < 0,1), pronóstico de talla adulta 146,5 cm (SDS 2,62). Rasgos fenotípicos de Turner (cuello alado, mamilas separadas). Genitales externos fenotipo femenino. Estadio puberal S1P1A1. Estudio hormonal: FSH 8,3 UI/l, LH 0,2 UI/l, 17-beta-estradiol < 20 pg/ml, testosterona libre (T) 0,09 pg/ml, T tras estímulo HCG 0,07 pg/ml, IGF1 155 ng/ml, IGFBP3 2,45 μg/ml, función tiroidea normal. Cariotipo mosaicismo 45 X/46 XY, la línea 46 XY en una proporción del 70-75 % y la línea 45 X en una proporción del 25-30 %. Ecografía pélvica: Utero de 3-4 cm de diámetro longitudinal (pequeño para la edad), no estructuras anexiales. IRM pelvis: No se visualizan ni utero, ni anejos. Estudio anatomopatológico: Gónada derecha: gonadoblastoma de 2 x 1 x 0,5 cm. Gónada izquierda: Tejido ovárico compuesto por estroma con ausencia de elementos germinales, trompas derecha e izquierda sin alteraciones histológicas significativas. Diagnóstico: DGM. Tratamiento: gonadectomía bilateral, tratamiento con hormona del crecimiento y terapia estrogénica sustitutiva en edad puberal.
Caso clínico 2: Neonato con genitales ambiguos. AP y AF sin interés. Exploración: genitales ambiguos, pene hipospádico con escroto hipoplásico, bífido y con rafe medio, no se palpan testes. Estudio hormonal: T basal 0,31 ng/ml, T tras estimulo HCG 3,9 ng/ml, dihidroT 0,77 ng/ml, LH y FSH < 0,1 UI/l, ACTH basal 25 pg/ml, cortisol basal 11 μg/ml, 17-OH-pregnenolona 0,43 ng/ml, 17-OH-progesterona 7 ng/ml, DHEAS 26 μg/dl, delta 4 androstendiona 8,6 ng/ml. Cariotipo: 46 XY. Genitografía: paso de contraste a vejiga, sin fístulas a otros órganos genitales, uretra corta. IRM pelvis: teste derecho de 2 cc en extremo superior de canal inguinal derecho, teste izquierdo de 1cc en anillo inguinal superficial. Anatomía patológica: Disgenesia testicular bilateral con restos mullerianos. Diagnóstico: SMD. Tratamiento: Biopsia testicular bilateral, fijación de teste derecho en escroto, gónada izquierda mínima e intrabdominal no se puede liberar. Se informa a los padres y acuerdan la decisión de conducirle como varón. Terapia de estimulación con testosterona 9 semanas y genitoplastia masculina.
Comentarios: La disgenesia gonadal parcial es una alteración de la diferenciación sexual, que implica un grupo heterogéneo de trastornos de la gónada y alteración del fenotipo. Los hallazgos clinicopatológicos en la DGM y SMD son similares. Esto sugiere que estas dos entidades son diferentes formas de la misma patología, por lo que se propone una única etiopatogenia. El desarrollo gonadal anormal es causado por aberraciones/mosaicismos o mutaciones/delecciones cromosómicas en el gen SRY o en otros genes responsables de la diferenciación gonadal. El diagnóstico diferencial anatomopatológico debe permitir clasificar a los pacientes con ambigüedad sexual entre DGM y SMD. El estudio histológico de estos casos es importante para un correcto diagnóstico y prevenir los cambios neoplásicos. Aunque lo habitual en estos pacientes es ser asignados al sexo femenino, la decisión sobre el sexo de crianza se debe basar en el potencial de función normal de los genitales externos. La alternativa de la reconstrucción masculina es una opción en caso de virilización importante y un teste descendido. En la pubertad los estudios hormonales informan de la capacidad funcional de los testes. Si hay una deficiencia la terapia hormonal es necesaria.
Variaciones fenotípicas en la presentación neonatal del seudohermafroditismo masculino
J.M. Caimari, J. Ferragut, M. Herrera, J. Bregante y J. Rossell
Pediatría. Hospital Universitario Son Dureta. Palma de Mallorca. Baleares. España.
Introducción: El seudohermafroditismo masculino engloba una serie de transtornos en la diferenciación sexual, debido a diferentes etiologías, entre ellas el síndrome de resistencia a los andrógenos (SRA) y el déficit de 5-alfa-reductasa. En época neonatal presentan genitales externos ambiguos o femeninos normales con presencia de gónadas masculinas y cariotipo 46 XY.
Casos clínicos:Caso 1. Recién nacida con genitales externos femeninos en cuya exploración se observa hipoplasia de labios menores y en labios mayores se palpan estructuras que parecen corresponder a gónadas. Es el segundo hijo de una madre sana, procedente de República Dominicana, con embarazo y parto normales. En la historia familiar hay varias hermanas de la madre con amenorrea y esterilidad. El estudio hormonal basal es normal. En la ecografía abdominal y RM no se visualizan útero ni ovarios y se observan gónadas en labios mayores. Estudio genético: cariotipo 46 XY. Estudio del gen AR revela una banda extra en el exón 4, cuya secuenciación muestra mutación D690G en hemicigosis. Evolución: al mes de vida ya no se detectan gónadas en labios; al 4.º mes se procede gonadectomía bilateral y herniorrafia inguinal derecha. El examen histológico revela tejido testicular neonatal normal. Casos 2 y 3: Recién nacidas gemelas, prematuras de 28 semanas de gestación (peso 890 g y 1.045 g). Madre sana sin antecedentes personales ni familiares de interés. A la exploración neonatal se aprecian genitales femeninos normales con aparición de hernias bilaterales a los 2 meses. Se solicita estudio ecográfico pélvico-abdominal: ausencia de genitales internos femeninos, probables testes en conducto inguinal. Test de HCG con cociente T/DHT normal. Estudio genético: cariotipo 46 XY. Estudio: mutación R607X en gen AR. Al año se efectua laparoscopia con gonadectomía bilateral y herniorrafia. Caso 4: Recién nacida normal, embarazo controlado, parto por cesárea, PAEG. Período neonatal normal. Antecedentes familiares sin interés. Remitido a los 23 días por aspecto atípico de genitales externos: edema de labios mayores, hipertrofia de clítoris y ausencia de orificio vaginal. No se palpan gónadas. Meato uretral en periné. Ecografía abdominal: no se visualizan genitales femeninos. Estudio hormonal tras HCG: índice T/DHT 18,6. Estudio genético: cariotipo 46 XY. Estudio molecular: mutación puntual en gen 5-alfa-reductasa tipo2. Evolución. A los 2 meses se palpa teste en labio mayor izquierdo y en conducto inguinal derecho, se practica gonadectomía bilateral al 6.º mes, con biopsia de piel genital. Caso 5: Recién nacida con diagnóstico prenatal de seudohermafroditismo masculino con cariotipo 46 XY y genitales externos femeninos. Antecentes familiares de dos hermanas de la madre con diagnóstico de SRA. Exploración genital neonatal normal. Ecografía abdominal: no se visualizan estructuras mullerianas. Estudio hormonal tras HCG: índice T/DHT 12,80. Estudio molecular del gen AR negativo, pendiente resultado del gen de 5-alfa-reductasa.
Conclusiones:1) Necesidad de una exploración genital detallada en el recién nacido. 2) La historia familiar detallada puede en ocasiones orientar el diagnóstico. 3) Utilidad de la ecografía como exploración inmediata. 4) El estudio genético es esencial para completar el diagnóstico. 5) Gonadectomía bilateral. 6) Necesidad de un equipo multidisciplinar para el tratamiento precoz, inmediato y a largo plazo.
Síndrome de insensibilidad a los andrógenos completa y sexo mental masculino
J.A. Bermúdez de la Vega, M.J. Villalobos Linares, M. López-Campos Bodineau y M. Jiménez Tejada
Servicio de Pediatría. Hospital Virgen Macarena. Sevilla. España.
Objetivo: Presentar un caso de insensibilidad complet a a los andrógenos (SIAC) asociado a sexo mental masculino.
Caso clínico:Anamnesis: Paciente de 13 años y 6 meses de edad, que coa desde siempre ser varón. Antecedentes personales: no de interés. Antecedentes familiares: amenorrea primaria en dos tías maternas y una prima hermana adultas. Tres hermanas de 10, 12 y 16 años con desarrollo sexual normal. Exploración física: vestimenta y ademanes masculinos. Genitales externos de aspecto femenino prepuberal. Ausencia de vello púbico (P1). No hipertrofia de clítoris. No palpación de testes inguinal. Vagina hipoplásica con fondo ciego a 2 cm. No desarrollo mamario (S1). Talla 154,9 cm. Peso 47,1 kg. Resto, sin hallazgos de interés. Exámenes complementarios: cariotipo 46 XY; test de LHRH (100 μg sc, 40 min): FSH/lH: 3/4,1, 14,8/53 mIU/ml; test de hCG (2,500 IU/48 h, 3 dosis), basales/postestímulo: testosterona total, 5/10,5 nmol/l; testosterona libre, 2,03/5,33 pg/ml; DHEA-s, 154/193 μg/dl; 17-OH-progesterona, 2,1/1,8 ng/ml; androstendiona, 4,76/3,86 ng/ml; androstanodiol, 1,9/5,8 ng/ml; dihidrotestosterona 0,080/0,110 nmol/l. Ecografía y RM abdominopélvica: no imágenes correspondientes a útero ni anejos, no testes intraabdominales. Edad ósea: 12 años (mujer), 13,4 años (hombre). Diagnóstico molecular: mutación concreta en el gen RA (cambio de C por G en el exón 2).
Comentarios: La incidencia de SIAC es de 1/20.000 recién nacidos con sexo genético masculino. Las mutaciones en el exón 2 suelen provocar formas completas. En éstas, el sexo que se asigna es el femenino, con adecuada adaptación psicosocial. El tratamiento consiste en la extirpación de testes y tratamiento hormonal sustitutivo desde la pubertad. El caso expuesto plantea una importante controversia terapéutica al asociarse a sexo mental masculino.
MISCELANEA
Evaluación clínica y genética del síndrome de Turner
T. Arévalo Saade, P. Cabanas, L. Castro-Feijoó, M. López, M. Fuster, J. Barreiro y M. Pombo
Unidad de Endocrinología Pediátrica, Crecimiento y Adolescencia. Hospital Clínico Universitario y Universidad de Santiago de Compostela. España.
Introducción: El síndrome de Turner (ST) es una enfermedad cromosómica causada por una completa o parcial monosomía del cromosoma X, con incidencia de 1/2.000-2.500 nacidos vivos, fenotípicamente niñas. Esta entidad nosológica está caracterizada por: rasgos dismórficos, disgenesia gonadal y en el 95-100 % una importante afectación de la talla constituyendo una de las indicaciones de hormona de crecimiento (GH).
Objetivos:1) Analizar las características clínicas del ST. 2) Mostrar los diferentes cariotipos reportados en estas pacientes. 3) Establecer la presencia de anomalías congénitas y demás enfermedades asociadas al ST.
Material y métodos: Estudio retrospectivo de pacientes diagnosticadas de ST (de enero de 1984 a diciembre de 2004). Se analizó: 1) Aspectos demográficos. 2) Características clínicas. 3) Cariotipo (FISH). 4) enfermedades asociadas. Para el análisis estadístico se utilizó el programa: SPSS 11.0.
Resultados: Fueron diagnosticadas 36 pacientes. La primera consulta se realizó a los 7,6 años ± 4,1 y el motivo de consulta más frecuente fue la talla baja. La edad media de diagnóstico fue 5,6 años ± 4,5. En el cariotipo (FISH) se evidencia: monosomía (45 X): 25 %, mosaicismo: 50 % y mosaico triple o complejo: 25 %. Los rasgos clínicos más frecuentes son: 1) Obstrucción linfática: Pterigium coli (51,6 %), cabello y pabellones auriculares de implantación baja (41,9 %), uñas hiperconvexas (25,8 %), linfedema de manos y pies (19,4 %). 2) Trastornos del crecimiento esquelético: talla baja (67,7 %), paladar ojival (74,2 %), cúbitus valgo (25,8 %) y acortamiento de 4.º metacarpiano. 3) Otros hallazgos clínicos: tórax ancho (61,3 %), mamilas lateralizadas (54,8 %), estrabismo (38,7 %), nevos (9,7 %) e hipoacusia (9,7 %). 4) Factores embriogénicos: anomalías renales (5,5 %) y cardiovasculares (16,7 %). La coartación de la aorta constituye la alteración cardiovascular más frecuente y el riñón en herradura, la renal. 5) Otros trastornos asociados: hipotiroidismo (11,1 %), osteoporosis (8,3 %), transaminasas elevadas (2,8 %) y carcinoma papilar de tiroides en 1 paciente.
El 94,4 % siguen tratamiento con GH, con edad media de inicio a los 8 años y en dosis de 0,3 mg/kg/día. Después de una media de 7 años de tratamiento, el 61 % alcanzó una talla final media de 146,9 cm.
Conclusiones: El ST puede no estar asociado a características clínicas distintivas, por lo que su diagnóstico puede ser pasado por alto. En nuestra revisión, el principal motivo de consulta así como el rasgo fenotípico más frecuente fue el retraso de crecimiento, sugiriendo la importancia del cariotipo en el estudio de la paciente con talla baja. La técnica de FISH permitió constatar que la mayoría de cariotipos Turner son mosaicismos.
Mutación del genAIREen el síndrome poliglandular tipo 1
M.ª M. Martínez López, E. Martín Campagne, I. González Casado, R. Álvarez Doforno, E. Delgado Cerviño y R. Gracia Bouthelier
Servicio de Endocrinología Pediátrica. Hospital Infantil La Paz. Madrid. España.
Introducción: El síndrome poliglandular autoinmune tipo 1 es una enfermedad autosómica recesiva que se caracteriza por aparición de al menos dos de los siguientes criterios: candidiasis mucocutánea crónica, hipoparatiroidismo autoinmune o insuficiencia adrenal primaria autoinmune. Se asocia a otros procesos autoinmunes: hepatitis, onicodistrofia, diabetes mellitus tipo 1, tiroiditis, hipopituitarismo, etc. Recientemente se ha descrito la asociación con una mutación en el gen AIRE (AutoImmune Regulator), situado en el cromosoma 21. Existen 45 mutaciones sin asociación entre las distintas mutaciones y la forma de expresión de la enfermedad. A continuación se presentan dos casos de mutación de gen AIRE estudiados en nuestro centro.
Caso 1: Niña de 4 años con crisis convulsivas. En el estudio completo sólo se objetiva hipocalcemia persistente (Ca total: 4,4 mg/dl; Ca iónico: 0,79 mmol/l) con hiperfosforemia (P: 10 mg/dl) y PTH indetectable, llegando al diagnóstico de hipoparatiroidismo de probable origen autoinmune. Se inicia tratamiento con calcitriol y calcio manteniéndose estable hasta los 9 años, en los que presenta hipocalcemia no controlada a pesar del aumento de aportes orales. Se diagnostica de cuadro malabsortivo de dudosa etiología infecciosa o inmunológica. A los 8 años y 6 meses: hipertransaminasemia y un año más tarde se diagnostica de hepatitis autoinmune. Cariotipo y estudio de CATCH 22 normales. Ante la presencia del hipoparatioidismo asociado a otros procesos autoinmunes, se sospecha un síndrome poliglandular tipo 1 y se solicita estudio genético: delección de 13 pares de bases en el exón 8 del gen AIRE (dominio PHD de la proteína). Es un punto caliente que se ha descrito en al menos 51 pacientes y es muy frecuente en Gran Bretaña, donde probablemente exista un efecto fundador. Los padres y al menos un hermano son heterocigotos. Actualmente tiene 11 años y se realiza seguimiento por si aparecen otros procesos.
Caso 2: Niña de 3 años y 9 meses con ictericia, coluria e hipocolia. Se detecta hiperbilirrubinemia (BT: 17,3 mg/dl y BD: 11,2 mg/dl), aumento de transaminasas y alteración de la coagulación. Tras descartarse otras etiologías se llega al diagnóstico de hepatitis de probable origen autoinmune. A los 4 años, estando asintomática, se detecta hipocalcemia (Ca total: 5,8 mg/dl y Ca iónico: 0,73 mmol/l) con hiperfosforemia (P: 8 mg/dl) y PTH indetectable llegando al diagnóstico de hipoparatiroidismo de probable origen autoinmune y se inicia tratamiento con calcio y vitamina D. A los 4 años y 2 meses: candidiasis oral, perineal y cutánea que responde a antimicóticos tópicos pero recidiva con su retirada. A los 10 años: anticuerpos antitiroideos elevados con función tiroidea normal. A los 11 años: distrofia ungueal. Desde los 4 años infecciones broncopulmonares de repetición con buen control con tratamiento médico. Ante la sospecha de un síndrome poliglandular tipo 1 se solicita estudio genético detectándose una mutación del gen AIRE pendiente de resultados definitivos. A los 16 años, espirometría con patrón obstructivo muy grave y bronquiectasias generalizadas con importante pérdida de volumen pulmonar. Se diagnostica posible bronquiolitis obliterante de probable origen autoinmune o infeccioso. Actualmente tiene 17 años y se encuentra en la UCI pendiente de trasplante pulmonar.
Macroprolactinoma. Aportación de un caso y revisión de la casuística de la SEEP en los últimos 5 años
B. Solís Gómez, M. Oyarzabal, M. Chueca, S. Berrade, A. Sola y G. Grau
Servicio de Pediatría. Unidad Endocrinología Pediátrica. Hospital Virgen del Camino. Pamplona. Navarra. España.
Introducción y objetivo: Los prolactinomas en pediatría son raros (2 % de los tumores intracraneales en niños y adolescentes, 50 % de los adenomas hipofisarios). Las últimas revisiones proponen la cabergolina como el tratamiento más eficaz y seguro, aunque hay pocos casos publicados en nuestro país. Por ello, hemos querido comunicar un caso recientemente diagnosticado en nuestra unidad, revisando al mismo tiempo los casos publicados en la SEEP en los últimos 5 años.
Caso clínico: Niña de 14 años, sin antecedentes personales ni familiares (menarquia a los 12 años, menstruaciones regulares). Acude por galactorrea bilateral de corta evolución. No refería tratamiento farmacológico alguno. Exploración física: Peso: 53.100 kg (P75). Talla: 166,4 cm (P90). Estadio de Tunner: 4. Galactorrea espontánea bilateral. Resto de la exploración física y neurológica, normal. Pruebas complementarias: Hemograma y bioquímica, sin alteraciones. Prolactina basal (PRL): 86,2 y 103,9 ng/ml, resto de hormonas hipofisarias normales. Resonancia magnética (RM) craneal: imagen nodular hipofisaria (10 x 6 mm), de aspecto quístico y parcialmente captante con gadolinio que no erosiona estructuras óseas. Tratamiento: Cabergolina, 0,25 mg/1.ª semana, continuando con 0,5 mg/2 veces a la semana. Evolución: A los 2,5 meses de tratamiento la niña consulta por cefalea y visión borrosa; destaca en la exploración del fondo de ojo un borramiento bilateral del borde nasal de la papila. Se realizó una campimetría, en la que no se apreciaron alteraciones, y una nueva RM que mostró una importante disminución del prolactinoma (4 x 4 x 2 mm). Las cifras de PRL se habían normalizado (0,5 ng/ml) y la galactorrea había desaparecido, atribuyendo los síntomas a la brusca disminución del adenoma.
Revisión: Desde el año 2000, se han comunicado en la SEEP 6 casos de macroprolactinomas, 2 niños y 4 niñas, con edades comprendidas entre 12 y 16 años. Los motivos de consulta fueron variados: cefalea (1), trastornos visuales (1), retraso puberal (2) y galactorrea (2). En 3 casos el tratamiento de elección fue bromocriptina (2 de ellos fueron además intervenidos), y los 3 restantes fueron tratados con cabergolina, obteniendo un reducción importante en el tamaño tumoral y una normalización de los valores de PRL en poco tiempo, con muy buena tolerancia.
Conclusiones:1) La cabergolina constituye un tratamiento eficaz y seguro en los prolactinomas, evitando en muchos casos la cirugía. 2) Constituye hoy en día el tratamiento de elección tanto en microprolactinomas como en macroprolactinomas. 3) Aunque sean poco frecuentes, es interesante la comunicación y revisión de los casos de prolactinoma con el fin de establecer pautas de seguimiento y tratamiento comunes.
Utilidad de monitorización continúa de glucosa para estudio de hipoglucemia en un paciente
R. Cebrián Rubio, M. Clemente, M. Gussinyer, E. Armengol y M. Losada
Unidad de Endocrinología Pediátrica. Hospital Universitario Vall d'Hebron. Barcelona. España.
Introducción: La hipoglucemia es motivo de consulta y estudio frecuente en pediatría. En este caso clínico se utilizó la monitorización continua de glucosa como elemento de ayuda diagnóstica.
Caso clínico: Paciente mujer de 10 años de edad remitida a consultas externas de endocrinología por presentar desde hacía 2 años episodios de palidez, sudoración e hipotonía sin pérdida de conocimiento relacionados con situaciones de estrés. No estaban relacionados con ejercicio físico ni ayuno. En alguna de estas ocasiones se realizó glucemia capilar, que fue < 45 mg/dl. Se había practicado electroencefalograma y registro Holter cardiológico, que fueron normales. Antecedentes familiares: sin interés. Antecedentes personales: Pretérmino de 28 semanas. Apgar 2. Peso al nacer 1.090 g. Cariotipo 46 XX con inversión pericéntrica en el cromosoma 9. Enfermedad de membrana hialina con posterior displasia broncopulmonar, múltiples ingresos por sobreinfecciones respiratorias. Hemorragia intraventricular grado III que siguió buena evolución. Atresia anorrectal intervenida a los 4 años. Cierre de colostomía a los 5 años. Atresia esofágica tipo I (sin fístula): a los 3,5 años intervenida mediante esofagoplastia con anastomosis termino-lateral esofagogástrica, vagotomía y piloroplastia. Tetralogía de Fallot con CIA ostium primum: corregida a los 2 años. Comunicación interventricular residual, insuficiencia pulmonar severa y tricuspídea moderada. No requiere tratamiento.
Exámenes complementarios: Monitorización continua de glucosa durante 3 días mediante el Continuous Glucose Monitoring System (CGMS), la paciente realizaba ingestas cada 4 h de alimentación espesa y ayuno nocturno de 12 h.
Resultados: 6 hipoglucemias nocturnas < 50 mg/dl. Hiperglucemias 130-150 mg/dl posprandiales seguidas a las 2 h de hipoglucemias < 50 mg/dl que sugieren de síndrome de Dumping por los antecedentes quirúrgicos de la paciente. Debido a las hipoglucemias nocturnas se ingresó a la paciente para estudio practicándose test de ayuno de 17 h: glucemia 48 mg/dl, insulina 3 μU/ml, cortisol 4 μg/dl. Tras ingesta se constató hiperglucemia de 228 mg/dl seguida a las 2 h de hipoglucemia de 40 mg/dl. Estudio hormonal: insulina 2,5 μU/ml, cortisol 2 μg/dl, ACTH 11 pg/ml.
Diagnóstico: Síndrome de Dumping asociado a déficit de ACTH.
Evolución: Se inició tratamiento con hidrocortisona en dosis de 12 mg/m 2 sc/día vía oral y se realizó segunda monitorización continua de glucosa durante 3 días; se observó desaparición de hipoglucemias nocturnas. Disminución de las hiperglucemias posprandiales a 120-140 mg/dl sin presentar hipoglucemia posterior.
Conclusiones: La monitorización continua de glucosa es un elemento de ayuda diagnóstica en el estudio de hipoglucemia y de evaluación de la respuesta al tratamiento. La asociación de síndrome de Dumping y déficit de ACTH no está descrita en la literatura, lo que dificulta su diagnóstico.
Lipodistrofia generalizada congénita en tres hermanos
J.M. Rial Rodríguez y M. López Méndez
Servicio de Pediatría. Hospital Nuestra Señora De Candelaria. Santa Cruz de Tenerife. España.
Introducción: La lipodistrofia congénita se caracteriza por la ausencia generalizada de tejido adiposo subcutáneo e intraabdominal desde el nacimiento o los primeros meses de vida, acompañada de resistencia a la insulina, hipertrigliceridemia y diabetes habitualmente no cetósica. Se han descrito dos formas clínicas de lipodistrofia congénita generalizada: La tipo 1, en que se conserva el tejido adiposo periarticular y de la órbita, con menor incidencia de miocardiopatía y de retraso mental, y diabetes menos precoz que la tipo 2. Presentamos la evolución clínica y el estudio genético molecular de tres hermanos con lipodistrofia congénita generalizada (síndrome de Berardinelli-Seip) tipo 2.
Caso 1: Paciente que es examinada por primera vez a los 4 meses de edad, presentando peso de 5.950 g (P20), longitud de 65,5 cm (P90). Se observa fenotipo característico con ausencia generalizada de grasa subcutánea, nalgas en bolsa de tabaco, mejillas hundidas y musculatura llamativa en las extremidades, con aumento de tamaño de las manos y los pies, y cabello rizado abundante. En la analítica realizada a la paciente destacan triglicéridos de 1.325 mg %, hiperoxaluria e hipercalciuria. La ecografía abdominal muestra hepatomegalia grasa. En la ecocardiografía se aprecia marcada hipertrofia septal asimétrica del ventrículo izquierdo con engrosamiento valvular. La paciente muestra un apetito exagerado que contrasta con la escasa ganancia ponderal. A la edad de 4 años sufre episodio de pancreatitis necrótico-hemorrágica coincidiendo con cifras marcadamente elevadas de triglicéridos. La diabetes aparece a los 9 años de edad. No se consigue reducir las cifras de triglicéridos pese a añadir medicamentos hipolipemiantes, con cifras de triglicéridos hasta 2.500 mg %. La paciente alcanza un desarrollo puberal completo y menarquia a los 13 años, siendo muy irregulares las sucesivas menstruaciones. La miocardiopatía evoluciona de forma muy lenta, desarrollando estenosis aórtica y pulmonar con escasa repercusión hemodinámica. La paciente tiene un rendimiento escolar deficiente en algunas áreas, pero alcanza el nivel normal en otras. La resonancia magnética de cerebro no detectó alteraciones hipotálamo hipofisarias ni dilatación del sistema ventricular. En el estudio neurofisiológico se constata neuropatía periférica moderada a la edad de 15 años.
Casos 2 y 3: Los dos hermanos varones nacen de gestación gemelar, parto a término con pesos al nacimiento de 2.535 g y 2.595 g y tallas de 51 cm y 52 cm. A la edad de 3 meses, muestran fenotipo lipodistrófico generalizado, con práctica ausencia de panículo adiposo en mejillas y tejido subcutáneo del tronco y las extremidades. Se constatan cifras elevadas de triglicéridos e insulina, sin hiperglucemia durante los dos primeros años. La ecocardiografía realizada a ambos es normal a la edad de 8 meses; se comprueba leve hipertrofia del ventrículo izquierdo a los 2 años. A los 2 años y medio se detecta hipoacusia neurosensorial en los dos gemelos, que presentan marcado retraso del lenguaje.
Análisis genético-molecular: El estudio genético fue realizado por el grupo investigador de Jocelyn Magré (INSERM, París), autores de la descripción del gen Gng3lg de la seipina. Los tres pacientes son heterocigotos compuestos para dos mutaciones del gen de la seipina, no encontrando alteraciones en el gen AGPAT-2. En el exón 2 presentan delección/inserción (536 del CCinsGGA) que provoca un cambio de pauta de lectura (frameshift) a partir del aminoácido 63 y la introducción de un codón de terminación en la posición 75. Esta mutación ha sido hallada en familias de Noruega, Reino Unido y Sicilia y en los casos presentados proviene de la línea paterna. En el exón 4 existe una inserción de adenina en posición 669 que ocasiona cambio en la pauta de lectura despues del aminoácido 108, con introducción de un codón de terminación en posición 113. Esta mutación proviene de la madre, y ha sido encontrada en familias de origen portugués residentes en Sudáfrica.
Prolactinomas en la adolescencia: heterogeneidad clínica y evolutiva
P. Casano Sancho 1, A. Ferrer, L. Ibáñez y C. Vall 2
1Endocrinología. 2Laboratorio Hormonal. Hospital Sant Joan de Déu. Esplugues de Llobregat. Barcelona. España.
Introducción: Los adenomas hipofisarios suponen menos del 2 % de los tumores intracraneales en la edad pediátrica, y los prolactinomas son los más frecuentes. Debido a su baja incidencia, la literatura en esta cohorte de edad es escasa y con pocos casos. La forma de presentación puede diferir de la de los adultos, donde predominan los transtornos menstruales como motivo de consulta en la mujer y la clínica neurológica en el hombre.
Objetivos:1) Conocer la forma de presentación del prolactinoma en la adolescencia. 2) Valorar su evolución clínica en esta población 3) Revisar la respuesta al tratamiento según parámetros clínicos, analíticos y de neuroimagen.
Sujetos y métodos: Estudio retrospectivo (enero de 2000-enero de 2005) de los pacientes menores de 18 años prepuberales o peripuberales diagnosticados de prolactinomas. Se recoge: historia clínica, exploración física (estadio puberal, Tanner) y neuroimagen (RM) al diagnóstico y cada 6 meses después del inicio del tratamiento; mediciones hormonales basales (PRL, TSH, LH, FSH) y postestímulo en los casos de sopecha de déficit hormonal asociado. Todos los pacientes recibieron carbegolina en dosis de 0,5-1,5 mg semanales.
Resultados: Presentamos 3 casos (2 varones y 1 mujer), de edad promedio al diagnóstico 14 años. El motivo de la consulta fue heterogéneo: amenorrea secundaria (n = 1), ginecomastia (n = 1), talla baja y pubertad retrasada (n = 1), y el período medio de evolución de la clínica fue de 2 ± 1 años. El diagnóstico se realizó por el hallazgo de concentraciones elevadas de prolactina (promedio, 80 μg/l; rango 17-1.386 μg/l). Todos presentaban en la RM imagen compatible con microadenoma (n = 2) o macroadenoma (n = 1). En todos se inicia tratamiento con carbegolina en dosis entre 0,5-1,5 mg semanales (promedio de 16 meses; 2 de ellos siguen en tratamiento en la actualidad). En el seguimiento se objetiva la disminución de los niveles de prolactina y del tamaño del prolactinoma, con normalización sólo parcial de la clínica; no se registraron efectos secundarios.
Conclusiones:1) El prolactinoma es un tumor infrecuente en la edad pediátrica con manifestaciones clínicas heterogéneas. 2) La variabilidad en su presentación puede demorar el diagnóstico e inicio del tratamiento. Diagnosticamos sólo la punta del iceberg ? 3) El tratamiento prolongado con cabergolina mejora parcialmente la clínica. Deberíamos utilizarlo siempre como primera opción terapeútica.
Ecografía pélvica en hiperandrogenismos durante la etapa posmenárquica
D. Martín Hernández, P. Prieto Matos, E. Vázquez Peñas, J. Cedeño Montaño y J. Prieto Veiga
Endocrinología Infantil. Hospital Universitario de Salamanca. España.
Introducción: Las consultas de niñas con clínica de hiperandrogenismos (anomalías menstruales, hirsutismo, acné, etc.) tras las primeras menstruaciones es cada vez más frecuente en las consultas de Endocrinología Pediátrica. El estudio que realizamos pretende valorar el interés de la ecografía pélvica en el diagnóstico etiológico; además se analizan otros parámetros de interés clínico-diagnóstico.
Pacientes y métodos: Se estudian 10 niñas diagnosticadas de hiperandrogenismo. Se analizan los siguientes datos: diagnósticos, edad de la primera consulta, edad de inicio de los síntomas, datos ecográficos, síntomas que motivan la consulta, datos antropométricos, fecha de la menarquia, estudio hormonal y tratamiento.
Resultados: Hiperandrogenismo ovárico funcional 7, hiperandrogenismo suprarrenal funcional 2, hirsutismo idiopático 1. Edad media de la consulta 14,46 años (12,1-17,5 años). edad de inicio de algún síntoma 9,35 años. Ecografía pélvica: Volúmenes medios de los ovarios, ovario derecho 9,18 ml (5,3-13,4 ml), ovario izquierdo 9,75 ml (4,5-120,4 ml), patrón de poliquistosis ovárica 4, teratoma ovárico 1, quiste ovárico 1, patrón ecográfico normal postmenárquico con volúmenes ováricos más grande de lo habitual en la mayoría de los casos. Signos/síntomas: hirsutismo 7, acné intenso 5, amenorrea 3, hipermenorrea 1, pelo muy graso 4, caída del cabello 4, estrías importantes 6, obesidad 2. Antecedentes familiares: madre diagnosticada de SOPQ, 2; DM2, 2; calvicies precoces en padres, 4; hirsutismos, 5. Datos antropométricos: talla valor medio + 0,l6 DE (0,63 + 0,8 DE). No se incluye un caso diagnosticado de displasia ectodérmica hipohidrótica que asociaba talla muy baja (2,87 DE). Peso valor medio + 0,5 DE, edad ósea- en la mayoría de los casos edad adulta, talla diana 159,8 cm, talla definitiva o pronóstico de talla adulta 163,2 cm. Edad media de las menarquias de las madre 13,2 años, edad media de las menarquias de las hijas 12,4 años. Una de las menarquias fue provocada por persistir a edades tardías amenorrea primaria. Estudio hormonal: test Leuprolide LH basal media 4,58 μU/ml, a las 3 h 13,6 mU/ml. FSH basal 5,88 μU/ml, a las 3 h 24 μU/ml. Cociente LH/FSH superior a 2 en todos los casos, Estradiol basal 53,9 pg/ml, a las 24 h 228,2 pg/ml. 17-hidroxiprogesterona a las 24 h 2,05 ng/ml (0,9-3,4 ng/ml). Valores superiores a 2 ng/ml en un 60 % de casos. test de ACTH: 17-OH-progesterona basal 0,98 ng/ml, tras estímulo, media de 2,77 ng/ml, cociente glucemia/insulina inferior a 4,5 en 7 casos.
Conclusión: La ecografía es una técnica de gran interés para el diagnóstico de hiperandrogenismos postmenárquicos. Es habitual encontrar ovarios más grandes de lo habitual y, en ocasiones, patrones de poliquistosis u otras ecoestructuras que ayudan al diagnóstico. La historia clínica, una correcta antropometría y el estudio hormonal permitirán llegar al diagnóstico definitivo.
Seudohermafroditismo masculino y alteraciones en el genWT-1
E. Martín Campagne, J. Guerrero Fernández, M. Álvarez-Acevedo, M.M. Martínez López, P. Lapunzina y R. Gracia Bouthelier
Servicio de Endocrinología Pediátrica. Hospital Infantil La Paz. Madrid. España.
Introducción: El gen WT-1 (gen del tumor de Wilms) está localizado en 11p13 y codifica un factor de transcripción con actividad supresora tumoral. En la embriogénesis actúa muy precozmente en el desarrollo renal y gonadal. Las anomalías de este gen son responsables de los síndromes de Denys-Drash (DDS), Frasier (FS) y WAGR. El DDS se caracteriza por la tríada de anomalías genitales, nefropatía (esclerosis mesangial difusa) y tumor de Wilms. El FS asocia disgenesia gonadal XY y nefropatía (glomeruloesclerosis focal y segmentaria). Los pacientes con FS no desarrollan tumor de Wilms, aunque a menudo presentan gonadoblastomas. El síndrome de WAGR cursa con tumor de Wilms (W), aniridia (A), anomalías genitourinarias (G) y retraso mental (R). Presentamos 2 casos clínicos relacionados con el gen WT-1, un síndrome de WAGR y un DDS.
Caso clínico 1: Neonato de 2 días de vida con aniridia bilateral y criptorquidia izquierda con pene de tamaño normal. Embarazo y parto sin incidencias, peso y longitud al nacimiento en percentil 25. Padres sanos, no consanguíneos. Pruebas complementarias: cariotipo 46 XY normal, ees evocados visuales normales. Evolución: intervenido a los 6 meses de hernia inguinal izquierda; se realizó orquidopexia y biopsia del teste criptorquídico. Anatomía patológica: disgenesia testicular con restos müllerianos. La asociación de aniridia y disgenesia testicular hace sospechar síndrome de WAGR, confirmándose en el estudio genético: deleción del gen WT-1 y del AN-2 o PAX-6: ish del(11) (p13p13)(AN-,D11S324-,WT1-). Actualmente, con 14 meses de edad, presenta retraso psicomotor moderado y alteraciones cerebrales difusas en la RM. Los controles ecográficos renales no han mostrado aún anomalías.
Caso clínico 2: Paciente de 1 mes con genitales externos ambiguos: micropene de 2 cm, hipospadias perineal y labios escrotalizados (Prader III). No se palpan gónadas. Embarazo y parto normales, peso y longitud al nacimiento en percentil 50. No antecedentes familiares de interés. Pruebas complementarias: cariotipo 46 XY normal; genitografía, RM y uretrocistoscopia: uretra masculina y utrículo prostático; laparoscopia: 2 testículos intraabdominales de pequeño tamaño sin evidencia de estructuras müllerianas; estudio genético (5-a -reductasa y receptores de andrógenos): normal. Evolución: a los 10 meses y medio palpación de masa abdominal, diagnosticándose por TC de probable tumor de Wilms que afecta los 2/3 inferiores del riñón. Se trata con quimioterapia y cirugía y se realiza biopsia testicular. Anatomía patológica: nefroblastoma rabdomiomatoso fetal, estadio 1. Restos nefrogénicos intralobares múltiples. Áreas de esclerosis mesangial difusa. Biopsia gonadal: disgenesia testicular y disminución del índice de fertilidad tubular. La presencia de la tríada característica hace muy probable el diagnóstico de DDS. Se confirma genéticamente mutación del WT-1: R366H (CGT > CAT).
Conclusiones: La presencia de nefropatía en un varón con seudohermafroditismo sugiere el diagnóstico de DDS o de FS, debidos a mutaciones del gen WT-1. El tumor de Wilms se desarrolla en > 50 % de los DDS, mientras que los niños con FS tienen riesgo de gonadoblastoma. El síndrome de WAGR, producido por microdeleción en 11p13 cursa característicamente con aniridia por mutación del gen contiguo PAX-6.
Macroorquidismo: a propósito de un caso
M. Álvarez-Acevedo, M.M. Martínez López, M.A. Molina Rodríguez, A. Oliver Iguacel, M. Nistal y R. Gracia Bouthelier
Endocrinología Pediátrica. Hospital Infantil La Paz. Madrid. España.
Introducción: El macroorquidismo es una entidad poco frecuente en niños. Puede encontrarse acompañado de retraso mental, orejas prominentes y alargamiento facial, constituyendo el síndrome de X frágil. Puede aparecer asociado a casos de hipotiroidismo severo de larga evolución, como resultado de un aumento de FSH independiente de la activación del eje hipotálamo-hipofiso-gonadal. Otras posibles causas de aumento de tamaño testicular son los restos adrenales en la hiperplasia suprarrenal congénita, los macroadenomas secretores de FSH y los linfomas En adultos el macroorquidismo bilateral (tamaño testicular > 25 cc) aparece en el 2 % de la población y generalmente constituye una variante de la normalidad. Se ha especulado acerca de una mutación en heterocigosis en el receptor de la FSH, que podría ser la causante de algunos casos de macroorquidismo.
Caso clínico: Varón de 11 años y 7 meses remitido al Servicio de Endocrinología Pediátrica por haber presentado en los últimos meses un llamativo aumento bilateral del tamaño testicular. Los antecedentes personales y familiares carecían de interés. En la exploración física se objetivó una pubarquia II-III y un volumen testicular izquierdo de 35 cc, y derecho de 30 cc, con un escroto que no presentaba hiperpigmentación. El pene se hallaba discretamente engrosado, siendo la longitud de 6 cm.
Pruebas complementarias:Sistemáticos de sangre y orina: normales. Determinaciones hormonales: FSH: 7,28 mUI/ml, LH: 1,21 mUI/ml, prolactina: 4,85 ng/ml, testosterona: 2,31 ng/ml, estradiol: 12 pg/ml, 17-OH-progesterona: 1,41 ng/ml. Marcadores tumorales: β -HCG: < 1 UI/ml, a fetoproteína: 1,74 ng/ml. Cariotipo: 46 XY normal. Ecografía testicular: testes de aspecto puberal de ecogenicidad homogénea y con vascularización normal. RX muñeca izquierda: correspondiente a una edad ósea de 11,5 años. Resonancia magnética cerebral con contraste: glándula hipofisaria de morfología normal sin asimetrías en la captación de contraste, tallo hipofisario centrado, estructuras paraselares sin alteraciones. Biopsia testicular: Parénquima testicular con maduración tubular puberal muy avanzada. El intersticio carece de células de Leydig de tipo adulto, observándose únicamente algunas células precursoras de aspecto fibroblástico. No se detecta lesión ocupante de espacio ni tampoco infiltrados inflamatorios. Evolución: Actualmente el paciente tiene 12,5 años. El tamaño testicular no ha variado respecto al objetivado en la primera consulta.
Conclusiones: Se trata de un caso de aumento exagerado del volumen testicular en los primeros estadios de la pubertad. Las determinaciones hormonales, el cariotipo, la ecografía testicular, la RM cerebral, y la biopsia son normales, lo que permite descartar las entidades hasta ahora descritas como causantes de macroorquidismo. Probablemente, como puede ocurrir en los adultos, se trate de una variante de la normalidad, aunque no se ha podido descartar la existencia de una mutación en el receptor de la FSH.
Hermafroditismo verdadero
M.V. Borrás Pérez, C. Piró 2, I. Anquela 1, J.C. Ferreres 2 y L. Audí 2
Servicio de Pediatría. 1Hospital de Granollers. 2Hospital Infantil Vall d'Hebron. Barcelona. España.
Introducción: El hermafroditismo verdadero (HV) es una forma de disgenesia gonadal que se caracteriza por la presencia simultánea de tejido testicular y ovárico con células germinales viables de ambas gónadas en un mismo individuo. Es muy poco frecuente, y la prevalencia real es difícil de precisar debido a la falta de diagnóstico histológico en muchos casos de seudohermafroditismo.
Caso clínico: Recién nacido de padres jóvenes no consanguíneos. No antecedentes de interés. Corresponde a una segunda gestación a término. Peso al nacer 3.990 g (P90); longitud: 51 cm (P75): perímetro craneal: 37 cm (P97). Genitales externos de fenotipo aparentemente masculino: Bolsa escrotal en el lado derecho con gónada palpable, bolsa residual izquierda similar a labio mayor femenino, pene de 2 cm longitud con hipospadias escrotal con chordee severa.
Exploraciones:Analítica hormonal (5 días de vida): 17-OH-progesterona: 1,5 ng/ml, testosterona: 1,1 ng/ml, D4-AT: 0,32 ng/ml, DHEA-S: < 15 μg/dl. Cariotipo en sangre periférica: 46 XX. Por técnicas FISH y PCR confirma la presencia de dos cromosomas X y ausencia del gen SRY. Ecografía abdominopélvica: Presencia de gónada de 1 cm de longitud con estructura y morfología compatible con testículo en bolsa escrotal derecha. Estructura tubular entre vejiga y recto con línea endometrial semejante a un útero. Cisto-genitografía: se observa el relleno de una estructura a 1-2 cm del cuello vesical que parece corresponder a una vagina corta y pequeña. Resonancia magnética: confirma la presencia de un teste en bolsa escrotal derecha y de una estructura retrovesical izquierda compatible con útero rudimentario.
Evolución: Se practica test HCG a los 2 meses de vida (3.000 U en 6 dosis) incrementando la testosterona de 1,6 ng/ml hasta 7,5 ng/ml sin observarse cambios en el tamaño del pene. De forma conjunta con los padres se decide la asignación de sexo femenino. Se practica genitoplastia feminizante: clitoridectomía parcial con resección de los cuerpos cavernosos y reconstrucción de los labios mayores Se extirpa la gónada derecha, que semeja un testículo y la gónada izquierda que recuerda un ovario junto con una trompa. Se biopsia el tejido genital para cultivo de fibroblastos y determinación de cariotipo. El estudio histológico reveló la presencia de tejido testicular y ovárico en ambas gónadas siendo la gónada derecha de predominio testicular y la izquierda mayoritariamente de tejido ovárico.
Diagnóstico: Hermafroditismo verdadero con ovotestes bilaterales.
Comentarios: El diagnóstico etiológico completo y correcto de las ambigüedades sexuales constituye un reto que requiere la colaboración de un equipo multidisciplinar. La asignación de un sexo civil no es una decisión fácil. En este caso se valoró para asignar sexo femenino la difícil corrección quirúrgica del hipospadias con buen funcionamiento posterior, la presencia de útero y vagina con cariotipo XX y la mayor posibilidad de función sexual normal como mujer en edad adulta.
Eficacia de los bisfosfonatos iv en la osteoporosis infantil
E.M. Barrios González, V. García Nieto 1, I. Rodríguez 2, J.M. Rial 2, M. López 2 y R. Cardona 2
Unidades de 1Nefrología y 2Endocrinología Pediátricas. Hospital Universitario Nuestra Señora de Candelaria. Centro de Salud Playa San Juan. Tenerife. España.
Introducción: Los bisfosfonatos son análogos sintéticos de los pirofosfatos que inhiben la resorción ósea por su acción sobre los osteoclastos, habiéndose demostrado en adultos su utilidad en el tratamiento de la osteoporosis. En niños sólo está aceptado su uso endovenoso, y existe poca experiencia con las nuevas moléculas más potentes como el ácido zoledrónico.
Material y métodos: Se presenta la evolución densitométrica de 6 pacientes varones de 9 ± 4,8 años (r: 2,3-14) con osteoporosis secundaria (4 osteogénesis imperfecta, 1 enfermedad de Duchenne, 1 osteoporosis idiopática juvenil) tratados dos con pamidronato iv (3 mg/kg/día) durante tandas de 3 días cada cuatro meses y cuatro con ácido zoledrónico (4 mg/1,73 m 2 de sc) una o dos dosis al año durante un tiempo de 1,84 años (r: 1-4). Se determinaron las concentraciones de osteocalcina, PTHi, 25OHD3, 1,25(OH)D, así como el cociente desoxipiridinolina creatinina en la segunda orina de la mañana. Se determinó la densidad mineral ósea antes y después del tratamiento con un densitómetro Hologic QDR 4500.
Resultados: En todos los casos la DMO mejoró. ZDMO previo al tratamiento 3,26 ± 0,73 (r: 2,15-4,04), ZDMO tras tratamiento 1,83 ± 1,43 (r: 3,64-0,47). La DMO fue significativamente mayor tras el tratamiento 0,5467 ± 0,14 frente a 0,3922 ± 0,09 g/cm 2 (p: 0,02). El cociente Dpyr/cr fue significativamente menor tras el tratamiento, 30,99 ± 20,37 frente a 64,13 ± 47,94 nmol/mmol (p: 0,04). La osteocalcina fue significativamente menor tras el tratamiento, 50,27 ± 18,95 frente a 83,31 ± 30,45 ng/ml (p: 0,02). Sólo en un caso se produjo una fisura ósea tras el inicio del tratamiento. En tres casos se encontraron efectos adversos: en uno fiebre, en otro hipofosfatemia sintomática con dolores óseos y musculares y el último con hipofosfatemia e hipocalcemia asintomáticas.
Conclusiones: Los bisfosfonatos i.v. reducen las fracturas y aumentan significativamente la DMO en pacientes pediátricos afectados de osteoporosis. Parámetros relacionados tanto con la resorción como con la formación ósea están disminuidos tras el tratamiento, reflejando una disminución del recambio óseo. Algunos pacientes presentan efectos secundarios de carácter leve. Se precisan estudios más amplios para conocer los beneficios y efectos secundarios a largo plazo de estos medicamentos en la edad pediátrica.
Asociación del síndrome del X frágil con fenómenos autoinmunes en una familia
P.P. Mialdea Valle, E. Gallego, J. Sánchez y G. Lledó
Sección de Endocrinología Pediátrica. Hospital Universitario 12 de Octubre. Madrid. España.
Introducción: aportamos en esta comunicación el caso de una familia en la que se han diagnosticado varias enfermedades autoinmunes, fundamentalmente alteraciones tiroideas y hepatitis, siempre en el sexo femenino. A partir de estos hallazgos, se realizó un estudio genético familiar que detectó en las personas afectadas dos tipos de alteraciones cromosómicas concomitantes: 46 XX, inv dup 9 (q12,q21) y cromosoma X frágil.
Personas y métodos: Se estudió a una familia española, de raza caucásica y residente en el área del Doce de Octubre. Composición y antecedentes personales: Padre (53 años): sano. Madre (38 años): ptosis palpebral unilateral congénita, hipertiroidismo, retraso mental leve. Primera hermana (7 años): retraso mental moderado, resto sin interés. Segunda hermana (6 años): retraso mental leve-moderado, resto sin interés. Tercera hermana (3 años): sana.
Resultados: En la primera consulta se valoró a la segunda hermana, en la que se había detectado previamente una hipertransaminasemia persistente. Se llegó al diagnóstico de hepatitis autoinmune tipo 1 tras un estudio por parte de Gastroenterología Pediátrica. Durante este proceso diagnóstico, se apreció un posible hábito hipertiroideo, por lo que se realizó una determinación de la función y de los anticuerpos tiroideos y se objetivó una elevación de T4 con supresión de TSH y positividad de los anticuerpos antitiroglobulina y antiperoxidasa. Con estos hallazgos es derivada a nuestra consulta, donde la anamnesis revela que, desde varios días antes de la detección de la hipertransaminasemia, había estado sufriendo un cuadro de astenia, prurito, palpitaciones, aumento del apetito y paradójica pérdida de peso; en ningún momento habían aparecido ictericia, hemorragias, temblores ni alteración de la conducta. También supimos que la paciente estaba afectada de un retraso mental leve-moderado, sin que aparecieran otras patologías relevantes en sus antecedentes. La exploración física era la siguiente: peso 19 kg (P25), talla 116 cm (P50-75), fenotipo peculiar (cabello fino, frente prominente, hipertelorismo) y aumento del volumen tiroideo, con un perímetro cervical de 26 cm; el resto de la exploración es normal. Las pruebas complementarias que se pidieron arrojaron los siguientes datos: anticuerpos TSI 22,2 UI/l (normal < 2), ecografía con un tiroides heterogéneo y aumentado difusamente de tamaño (aunque sin nódulos), cariotipo 46 XX inv dup 9 (q12,q21) y estudio de X frágil con un cromosoma normal y otro con un número claramente patológico de repeticiones. Ante estos hallazgos, se decide estudiar al resto de la familia. La primera hermana presentaba una exploración normal (salvo por un fenotipo peculiar parecido) y positividad para los anticuerpos antitiroideos, con función y TSI normales; la ecografía era anodina. Se diagnosticó de tiroiditis eutiroidea y se realizó estudio genético, en el que se encontraron las mismas alteraciones genéticas que en la probanda. La tercera hermana solamente compartía el fenotipo, siendo el resto de la exploración y de las analíticas normales. El resultado de su estudio genético señaló también las mismas dos alteraciones cromosómicas. Por su parte, en el padre no se obtuvieron datos patológicos, y era sano en todos los aspectos. Y en la madre, que compartía fenotipo y retraso mental con sus hijas, también se encontraron las anomalías 46 XX, inv dup 9 (q12,q21) y X frágil.
Discusión: En la familia objeto de nuestro estudio, las alteraciones autoinmunes parecen razonablemente asociadas a una de sus alteraciones cromosómicas, o a una combinación de las dos. Estos fenómenos de asociación son conocidos en otras cromosomopatías, especialmente en el síndrome de Down. Sin embargo, nuestro caso resulta una novedad puesto que no están descritos para la duplicación del segmento (q12,q21) del cromosoma 9 (la inversión aislada es asintomática, y los que están descritos afectan fundamentalmente a otros segmentos) ni para el síndrome del X frágil. Sería recomendable, por tanto, realizar más estudios que pudiesen confirmar esta propuesta de asociación.
Hiperplasia virginal bilateral de la mama
M.P. Gutiérrez Díez, P. Flores Pérez, B. Rubio Gribble, J. García Domínguez, M.J. Galobardes y M.E. Corrales
Pediatría. Hospital Universitario de Getafe. Madrid. España.
Introducción: El primer caso de hipertrofia mamaria juvenil o hipertrofia virginal fue publicado por Durston en 1669. Se trata de un síndrome poco común caracterizado por el crecimiento exagerado unilateral o bilateral de la mama al inicio de la pubertad. El desarrollo de la glándula está regulado por múltiples hormonas y factores de crecimiento (estrógenos, PRL, GH, insulina, IGF-I, glucocorticoides y hormonas tiroideas). A pesar de ello, los intentos de control con tratamientos endocrinos habitualmente no han tenido éxito en las series publicadas, por lo que se ha recurrido a la cirugía reductora.
Caso clínico: Paciente de 12 años de edad que acude a consultas externas por crecimiento rápido de ambas mamas de 2 meses de evolución que se inicia tras la menarquia. Este crecimiento mamario ha modificado su ritmo habitual de vida, no acude al colegio y ha adoptado una conducta de aislamiento social. Refiere dolor intenso en la espalda y en ambas mamas, molestándole incluso el roce de la ropa. En la exploración física pr esenta unas mamas de gran tamaño, de color cianótico, con circulación colateral muy llamativa en tercio superior, de consistencia dura y con zonas de piel macerada, que afectan también a la areola y el pezón. Estadio de Tanner P3 A3. No es obesa. Resto de la exploración normal, excepto marcada cifosis dorsal. Los estudios analíticos y hormonales no revelaron ninguna alteración. La ecografia de mamas mostraba un parénquima hiperecogénico con múltiples imágenes anecoicas compatibles con quistes, senos lactíferos prominentes y conductos dilatados. En la punción biópsica, patrón anatomopatológico típico: exagerada proliferación del tejido conectivo asociado a una hiperplasia significativa del componente glandular. Por la maceración de la piel, la mal irrigación de las mamas y el dolor se decide la colocación de un vendaje compresivo hasta su reducción quirúrgica.
Discusión: La hiperplasia virginal suele presentarse antes de la menarquia o muy próxima a ella, como ocurrió en nuestra paciente, con un crecimiento rápido que puede afectar a una o a las dos mamas. Hay varias hipótesis sobre su etiología: respuesta exagerada del tejido mamario a los cambios normales de estrógenos circulantes durante la pubertad, alteración de las vías hepáticas de glicuroconjugación implicadas en la desactivación hormonal, etc. Más recientemente se ha propuesto la intervención de factores autoinmunes que interferirían con el complejo receptor mamario o la presencia en el suero de otros factores, distintos a los hormonales, que estimularían la hipertrofia del órgano. Histológicamente se caracteriza por una hipertrofia del estroma con degeneración quística de los conductos lactíferos, edema intersticial y la presencia de lóbulos de grasa. Debe hacerse el diagnóstico diferencial con otras causas de gigantomastia puberal: hipertrofia gestacional, inducción por drogas, enfermedad de Cowden o la presentación atípica de algunas leucemias o linfomas. El tratamiento de elección es quirúrgico, mamoplastia reductora o submastectomía subcutánea. La mamoplastia mejora los resultados estéticos pero no elimina completamente la posibilidad de recurrencias. Se han hecho múltiples intentos de control del crecimiento prequirúrgicos y posquirúrgicos con terapias hormonales: tamoxifeno (ha conseguido resultados temporales pero parece perder su efecto a largo plazo); acetato de medroxiprogesterona (inhibidor de la LH y efecto antiestrogénico directo, a largo plazo su uso se relaciona con el desarrollo de nódulos mamarios benignos y aumento del riesgo de aparición de neoplasias); bromocriptina (no ha sido efectiva; las concentraciones séricas de prolactina en estas pacientes son normales en la mayoría de los casos, aunque pueden estar más elevadas en los casos más severos).
Conclusión: La hiperplasia mamaria juvenil es un trastorno poco común, de etiología aún no aclarada. En los casos graves está indicada la cirugía reductora, aunque conlleva riesgo de cicatrices queloideas y disminución o falta de sensibilidad en la areola. Se puede asociar el tratamiento con tamoxifeno o medroxiprogesterona, asumiendo efectos secundarios como amenorrea o aparición de nódulos, pero serían necesarios más estudios de los receptores mamarios y sus ligandos, para mejorar las alternativas terapéuticas hormonales.
Complicaciones endocrinas de los tumores hipotalámicos infantiles
L. Ruiz Pérez 1, M. Zapico Álvarez-Cascos 1, C. Moscardó Guilleme 1, C. Esquembre Menor 2 y M. Tasso 2
1Unidad de Endocrinología Infantil. 2Servicio de Oncología Infantil. Hospital General Universitario de Alicante. España.
Introducción: Los tumores cerebrales son el tipo más frecuente de tumores sólidos en la infancia. La supervivencia de los mismos ha aumentado en la última década. Sin embargo, la mejora del pronóstico ha supuesto un aumento en el número de secuelas a largo plazo tras el tratamiento. Los efectos secundarios tardíos endocrinológicos son frecuentes y en muchas ocasiones van a ser tributarios de un tratamiento hormonal sustitutivo. Destacan las alteraciones de crecimiento, disfunción tiroidea, diabetes insípida, alteraciones función gonadal y de la pubertad.
Material y métodos: Se estudiaron 8 pacientes diagnosticados de tumores zona hipotalámica (3 craneofaringiomas y 5 gliomas de zona quiasma-hipotalámo) con secuelas endocrinológicas. Un total de 4 pacientes recibieron sólo tratamiento quirúrgico, 2 cirugía + quimioterapia, 1 cirugía + radioterapia, y 1 cirugía + quimioterapia + radioterapia.
Resultados: Se observó déficit de GH en 4 niños, alteración eje suprarrenal en 3 niños, alteración hipotiroidea en 3 niños, diabetes insípida en 4 niños, pubertad precoz en 2 niños e hipogonadismo 2.º-3.º en 1. Todos ellos han presentado buena evolución clínica y analítica con tratamiento hormonal sustitutivo.
Conclusiones: Todos los pacientes supervivientes de un tumor pediátrico deben ser vigilados periódicamente desde un punto de vista endocrinológico, al objeto de detectar precozmente la presencia de alteraciones hormonales. Es necesario realizar una valoración auxológica, hormonal y de fertilidad, tratando las posibles deficiencias hormonales, vigilando el desarrollo de la pubertad y analizando las posibles alteraciones en la fertilidad derivadas del tratamiento quirúrgico, radioterápico y quimioterápico que reciben estos pacientes.
