



18, 19 y 20 de mayo de 2006, Murcia
Evaluación de la pubertad y la función gonadal en pacientes pediátricos supervivientes de un cáncer infantil
I. Martín Ibáñez1, O. Cruz Martínez2, A. Parareda2, J. Mora2, J. de Torres Estella3, C. Valls4 y C. Pavía5
1Servicio de Pediatría. 2 Sección de Oncología. 3Sección Hematología. 4Servicio de Bioquímica. Hospital Universitario Sant Joan de Déu. Universidad de Barcelona. 5Servicio de Endocrinología. Hospital de Nens. Barcelona. España.
Objetivos: La mayor supervivencia en el cáncer infantil condiciona una mayor preocupación por los efectos tardíos del cáncer y su tratamiento. El objetivo es ampliar el conocimiento de las secuelas sobre la pubertad y la función gonadal en los supervivientes del cáncer infantil.
Pacientes y métodos: Se definieron un grupo de estudio y un grupo control. Grupo de estudio: pacientes con antecedente de cáncer infantil, con 2 años o más en remisión y sin tratamiento y edad entre 7 y 18 años al iniciar el estudio. Se seleccionaron 126 pacientes (72 niños y 54 niñas). Grupo control: niños sanos de 7 a 18 años (58 niños y 56 niñas). Métodos clínicos: curva de crecimiento, desarrollo puberal según Tanner, volumen testicular y ciclo menstrual. Métodos analíticos: determinación de concentración sérica basal de FSH, LH, testosterona, estradiol e inhibina B. En varones del grupo de estudio menores de 15 años se propuso un seminograma, y ecografía pélvica para niñas puberales.
Resultados:Grupo de estudio: edad media al diagnóstico 4,93 años, media de 6,53 años desde el fin del tratamiento. Diagnósticos: leucemia (43 %), HCL (10 %), LNH (8 %), Wilms (7 %), tumores SNC (6 %). Tratamientos: quimioterapia 97,6 % de los pacientes, radioterapia 26 %, neurocirugía 6 %, corticoides 75 %. Desarrollo puberal: 6 % de varones con retraso puberal y 15 % de niñas con pubertad adelantada (13 %) o precoz (2 %). Seminograma: efectuado en 10 varones (2 azoospermias, 2 astenozoospermia, 1 aspermia). Ecografía pélvica normal en 35 de 38 niñas. Comparando grupo de estudio frente a grupo control: los varones en estadio I y II tenían una edad significativamente mayor. En el estadio II FSH, LH y testosterona fueron significativamente mayores (2,71 UI/l frente a 1,55 UI/l, 2,00 UI/l frente a 0,90 UI/l y 6,2 nmol/l frente a 1,6 nmol/l), y en el estadio III se mantenían las diferencias significativas para LH (3,20 UI/l frente a 1,68 UI/l) y testosterona (12,9 nmol/l frente a 4,7 nmol/l). En las niñas no había diferencias en las variables de perfil, pero en el estadio I la inhibina B era significativamente menor en el grupo de estudio (< 10 pg/ml frente a 21 pg/ml). En 19 varones (26,4 %) existía insuficiencia tubular, asociada significativamente con la edad al inicio del tratamiento (×1,226 por año) y con el trasplante de médula ósea (TMO), y en 5 (7 %) insuficiencia de células de Leydig, asociada significativamente con la radioterapia (×5,663) y TMO. Entre las niñas, 11 (20,4 %) tenían insuficiencia ovárica parcial, asociada significativamente con la radioterapia (×13,407), la quimioterapia gonadotóxica (×9,096), el TMO y los tumores del SNC.
Conclusiones: La edad prepuberal al inicio del tratamiento no protege del daño gonadal. Todos los niños tratados de un cáncer deben ser vigilados durante la pubertad, ya que desde la prepubertad y etapas precoces del desarrollo pueden aparecer hallazgos clínicos y analíticos sugestivos de insuficiencia gonadal. No hay un marcador clínico, hormonal o ecográfico que sea suficiente para determinar si existe insuficiencia gonadal, lo que obliga a que el seguimiento de estos niños se prolongue a la edad adulta.
Nuevas técnicas diagnósticas: impacto clínico de la PET-FDG en oncología infantil
F. Rodríguez-Argente1, O. Cruz1, J. Mora1, A. Parareda1, C. de Torres1, J.R. García2 y M. Simó2
1Hospital Sant Joan de Déu. 2CETIR, Unitat PET. Barcelona. España.
Objetivo: Valorar el impacto de la PET-FDG en el tratamiento de los pacientes pediátricos oncológicos.
Pacientes y método: Se realizaron 56 estudios con PET en 38 pacientes (2-18 años), con diagnósticos de: enfermedad de Hodgkin (21 PET), linfoma no hodgkiniano (n:13), sarcoma (n:10), neuroblastoma (n:5), astrocitoma (n:3), histiocitosis (n:3), meduloblastoma (n:1). El motivo de la PET-FDG fue: sospecha de recidiva o diagnóstico inicial (n:17), control de la terapia (n:39). El estudio PET-FDG se realizó en un equipo GE.Advance.Nxi (6 MBq/kg). Tras el PET se determinó el estadio de la enfermedad y se analizó el manejo terapéutico.
Resultados: En 13/17 estudios (76,5 %) por recidiva la PET mostró lesiones hipermetabólicas compatibles con enfermedad activa: 11 verdaderos positivos (VP), 1 falso positivo (FP) en un LNH con captación de FDG en mediastino anterosuperior izquierdo y biopsia de rebote tímico, 1 VP-FP (neuroblastoma, PET con recidiva suprarrenal, pero con captación de FDG en hilio pulmonar derecho y evolución clínico/radiológica de neumonía). De los 6/17 casos (35,3 %) con PET negativo únicamente en dos se objetivo recidiva tumoral: 6 VN, 2 FN (1 astrocitoma y 1 meduloblastoma con progresión local). En 29/39 estudios (74,4 %) para valoración evolutiva al tratamiento la PET no evidenció enfermedad activa, confirmada por seguimiento: 29 VN. En 10/39 (34,5 %) la PET mostró persistencia de captación de FDG: 8 VP, 2 FP (enfermedad de Hodgkin, con persistencia de captación mediastínica y neuroblastoma, con captación en tibia; ambas sin evidencia de enfermedad por seguimiento). En total, en 20/56 casos (35,7 %) la PET motivó la realización de tratamientos adicionales: confirmación de recidiva (n:12), persistencia de enfermedad postratamiento (n:8).
Conclusiones: La indicación más frecuente de PET-FDG en nuestro ámbito fue el linfoma. La PET-FDG muestra un importante impacto en oncología pediátrica, tanto en la sospecha de recidiva como para el control del tratamiento. En un número significativo de casos afecta al manejo terapéutico.
131I-MIBG terapéutica en neuroblastoma de alto riesgo: experiencia de nuestra unidad de oncología infantil
O. Escobosa Sánchez, A. Herrero Hernández y T. Acha García
Unidad de Oncología Infantil. Hospital Materno-Infantil Carlos Haya. Málaga. España.
Objetivo: El grupo de pacientes con neuroblastoma (NB) de alto riesgo tienen una pobre respuesta a la terapéutica habitual, con una supervivencia global a los 5 años en torno al 20-30 %. Por este motivo, existe un marcado interés en la búsqueda de terapias más efectivas. Está demostrado el efecto de la radiación dirigida a las células tumorales mediante el empleo de 131I-MIBG terapéutica. Existen pocos estudios que muestren el manejo y posterior seguimiento y evolución de los pacientes sometidos a esta terapia. Presentamos nuestra experiencia con el manejo de este tipo de terapéutica.
Material y métodos: Estudio retrospectivo de los pacientes con NB de alto riesgo tratados en nuestra unidad con 131I-MIBG entre los años 1981 y 2006. Se revisan datos epidemiológicos, anatomopatológicos, tipo de terapia previa, situación clínica del paciente antes de la administración del radionúclido, dosis total y por kilogramo empleada, número de ciclos, el empleo de quimioterapia asociada, la necesidad de rescate con progenitores de sangre periférica, la toxicidad constatada y la situación actual de los pacientes.
Resultados: En los últimos 25 años han sido diagnosticados y tratados en nuestra unidad un total de 91 pacientes pediátricos con neuroblastoma, perteneciendo al grupo de alto riesgo un total de 17 (todos estadio IV) (18,7 %), a los que se administró terapia intensiva según el protocolo de tratamiento vigente. En cinco de estos pacientes se ha empleado terapia con 131I-MIBG.
Se trata de 4 varones y una niña, con edades comprendidas entre 2,3 y 14 años al diagnóstico, todos estadio IV, con tumor primario suprarrenal, paravertebral o torácico y metástasis en médula ósea en todos los casos, y en hígado u óseas en 2 pacientes. La histología mostraba en tres de ellos un NB pobremente diferenciado y en dos, ganglioneuroblastoma. Solamente un paciente presentaba amplificación del n-Myc, y en dos existía la deleción 1p. Los pacientes recibieron diferentes regímenes de quimioterapia, en función del protocolo SEOP o SIOP vigente en el año de diagnóstico, y al momento de emplear la 131I-MIBG terapéutica se encontraban en situación de enfermedad progresiva. La dosis de 131I-MIBG oscila entre 10 y 20 mCi/kg de peso, administrándose en 1 o 2 ciclos, separados entre sí desde 16 días hasta 2,8 meses, con bloqueo tiroideo. Dos pacientes recibieron quimioterapia asociada. El principal efecto adverso asociado a este tipo de tratamiento es la trombocitopenia, que fue más intensa en 2 pacientes y con una mediana de duración de 17 semanas. En 2 pacientes se realizó rescate con progenitores de sangre hematopoyética. Un paciente desarrolló leucemia no linfoblástica aguda en relación con la dosis total acumulada de VP-16. No se ha constatado toxicidad tiroidea o cardíaca a largo plazo. La situación actual de este grupo de pacientes es de dos vivos con enfermedad, y tres fallecidos, por progresión de la enfermedad o por el desarrollo de una leucemia mieloblástica aguda secundaria en un caso.
Conclusiones: La terapia con 131I-MIBG es un arma más en el tratamiento del NB de alto riesgo en pacientes pediátricos. Se considera una de las terapias que permiten cronificar la evolución de los NB-IV, mejorando así su supervivencia global. Su empleo está al alcance de algunas unidades de oncología infantil, con colaboración con el servicio de medicina nuclear. Es una terapia en general bien tolerada, cuyo principal efecto secundario es la trombocitopenia transitoria.
Infecciones fúngigas invasivas en una unidad oncopediátrica
M.B. Fernández, N. Dopazo, M. Dieste, C. Calvo, A. Carboné y A. Muñoz
Unidad de Oncopediatría. Hospital Infantil Miguel Servet. Zaragoza. España.
Objetivos: La frecuencia e incremento de infecciones fúngicas invasivas en pacientes hemato-oncológicos de alto riesgo, su elevada mortalidad y difícil tratamiento, nos ha llevado a analizar la mejor evolución de nuestros pacientes con el uso de los nuevos antifúngicos.
Material y métodos: Análisis retrospectivo de 13 casos de infecciones fúngicas invasivas en nuestra unidad (enero de 1994 a diciembre de 2005). Variables: diagnóstico de base, fase del tratamiento, germen aislado, lugar de aislamiento, pruebas de imagen patológicas, tratamiento y evolución.
Resultados: (tabla 1)
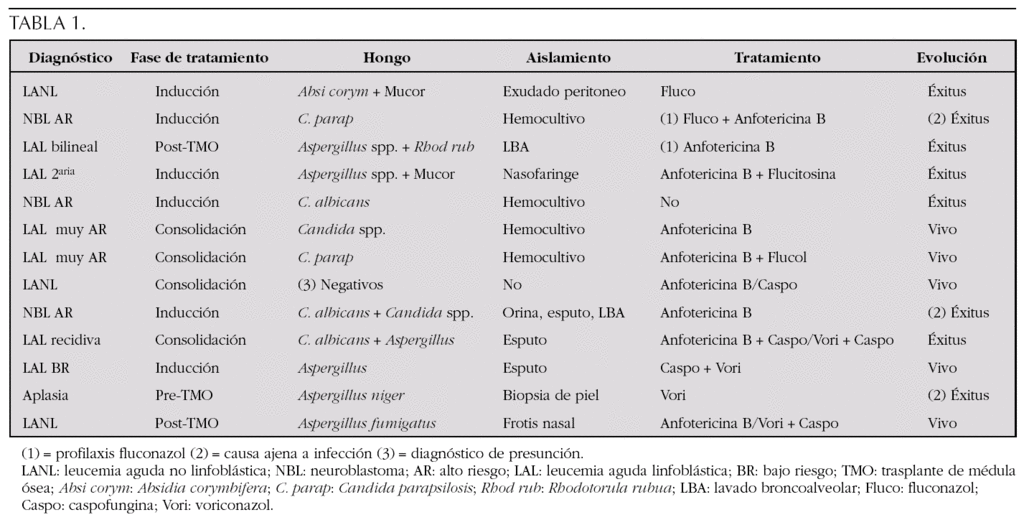
De las 6 candidiasis, 4 fallecieron (2 por causa ajena a la infección, 1 por aspergilosis sobreañadida); todos ellos recibieron tratamiento con anfotericina B, excepto el caso asociado a aspergillus. De los 6 casos de aspergilosis, fallecieron 4 (1 por causa ajena a la infección, ya que ésta se había resuelto con Voriconazol). Del resto de casos fallecidos por aspergilosis, sólo 1 había recibido voriconazol, como tratamiento de rescate.
Conclusiones: 1. En los últimos años ha habido un incremento en el número de casos de aspergilosis. 2. Las candidiasis responden bien al tratamiento con anfotericina B. 3. El uso de los nuevos antifúngicos ha mejorado el pronóstico (evolución y supervivencia); su inicio debe ser precoz.
Retinoico en NB, ¿hasta cuándo?
I.F. Canales, A. Teijeiro, I. Astigarraga, M.A. Piñán y A. Navajas
Hospital Cruces. Baracaldo. Bilbao.
Introducción: El NB (neuroblastoma) IV en niños mayores de un año sin factores biológicos desfavorables cursa con supervivencia No superiores al 30 %. Existe evidencia de prolongación en la supervivencia en algunos casos con el uso de ácido retinoico (AR), aunque a largo plazo se desconocen los efectos adversos. Este es el caso de un NB IV que lleva actualmente 7 años de supervivencia con tratamiento prolongado de AR.
Caso clínico: Varón de 14 años diagnosticado alos 7 de NB IV que consultó por dolores intermitentes de 1 año de duración, fiebre y anemia. Como datos positivos destacar al diagnóstico: NSE (enolasa): 188, LDH 2.055, ferritina 496 y dopamina en orina 920. En la TC: tumoración retroperitoneal de 3,7* 2,7 cm con metástasis ganglionares retroperitoneales, mesentéricas y mediastínicas. Infiltración masiva (92 %) en M.O. metástasis óseas múltiples (MIBG). Inmunohistoquímica: 0-3 copias de amplificación NMYC. Se inició tratamiento con quimioterapia a altas dosis, exéresis parcial (10/99), TASP (11/99), RT abdominal (12/99) ante persistencia captación por la masa, AR (6 + 6 tandas, hasta 1/01) y segunda cirugía por resto positivo en MIBG. En evalución fin de tratamiento (3/00) se evidencia captación pélvica, recibiendo por ello 131MIBG por 3 tandas de 100 mciI. En 1/2 se observa recaída en M.O. por lo que se administra quimioterapia (CFM y TPT), consiguiendo una respuesta parcial (MIBG + en huesos). En 10/02 se da TMZ (8 ciclos) y se consigue buena respuesta: MIBG negativa (2/03). Dado el alto riesgo se reinicia AR (12/03. 6 ciclos). Nuevamente en control de 5/04 se observa recaida en RM: metástasis óseas, por lo que recibe quimioterapia con CFM y TPT. Ya en el siguiente control del 5/05, la MIBG es . Se reinicia AR y debe ser suspendido por hipertrigliceridemia (hasta 639). tres meses más tarde es diagnósticado de hipotiroidismo, se inicia tratamiento sustitutivo y desde entonces lleva 7 meses con AR a dosis reducida al 75 % y sin problemas. En estudio de enfermedad mínima residual (EMR) realizado en seguimiento presenta fluctuaciones de TH y anti-GD2 en MO y sangre periférica que se negativizan durante la administración de AR y TPT.
Conclusiones: Observamos en este caso la supervivencia prolongada a costa de importantes efectos secundarios: hipotiroidismo (tratamiento con MIBG), hiperlipidemia mixta (AR e Hipotiroidismo), hemocromatosis con ferritina de 1942 y ecografía hepática compatible, repetidas hospitalizaciones, hasta 20. La adaptación y percepción de la familia ha sido un factor muy importante. Actualmente cursa 3.º de E.S.O. y está socialmente muy bien adaptado. Desconocemos cuál habría sido la evolución sin tratamiento modulador.
Tratamiento y profilaxis secundaria con voriconazol en un paciente pediátrico con lla y aspergilosis pulmonar
J.J. Úriz Monaut1, N. García de Andoin Barandiaran1, P. Esparza Paz2, C. Calvo Monge2, J. Landa Maya2, A. Vivanco López2 y R. Guerrero Pereda1
1Unidad de Hemato-Oncología Pediátrica. Servicio de Pediatría. 2Unidad de Cuidados Intensivos Pediátricos. Servicio de Pediatría. Hospital Donostia. San Sebastián. España.
Introducción: Aunque la eficacia y seguridad de voriconazol para el tratamiento de infecciones fúngicas en niños ha sido demostrada en los últimos años, su uso como profilaxis secundaria para prevenir la reactivación de dichas infecciones presenta pocas referencias en la literatura médica limitándose a series cortas en adultos.
Caso clínico: Presentamos un caso de una niña de 3 años y 6 meses con fiebre alta y malestar general en cuya exploración física se aprecia palidez marcada, petequias y hepatoesplenomegalia. Analítica: neutropenia grave (50/μl), plaquetopenia (16.000/μl) y anemia (8 g/dl). El aspirado medular reveló el diagnóstico de LLA pre-B, L2 de la FAB. Fue tratada con con el protocolo PETHEMA-2001 de bajo riesgo comenzando el ciclo de inducción con prednisona a 60 mg/m2/día y triple terapia intratecal (metotrexato, hidrocortisona y citarabina) continuando con asparraginasa intramuscular vincristina intravenosa, daunorubicina intravenosa y ciclofosfamida intravenosa a partir del octavo día de ciclo. Precisó antibioterapia (cefepima 13 días) por neutropenia febril, con cultivos negativos. El día 21 nuevo episodio de fiebre persistiendo neutropenia tratándose con cefepima y vancomicina intravenosa cediendo la fiebre al cuarto día. A partir del día 28 empeoramiento progresivo con cuadro de dolor y distensión abdominal, febrícula mantenida y deposiciones dispépticas sin sangre. La ecografía abdominal fue compatible con tiflitis precisando nutrición parenteral, analgesia con cloruro mórfico, antibioterapia (imipenem y vancomicina), terapia antifúngica (anfotericina B liposomal), soporte hemodinámico (dopamina), y G-CSF. El día 30 aumento de PCR y de necesidades de oxígeno con radiografía de tórax con tenue infiltrado intersticial bilateral añadiéndose a tratamiento cotrimoxazol y eritromicina. El día 32 episodio de hemorragia en cavidad oral con plaquetopenia que precisa transfusión de plaquetas y cauterización en quirófano, pudiéndose extubar 24 h después. Tras 2 días se aísla en tubo endotraqueal Aspergillus fumigatus, sustituyéndose anfotericina B liposomal por voriconazol intravenoso (6 mg/kg/12 h seguido de 4 mg/kg/12 h a partir del segundo día). La TC posterior mostró afectación alveolar difusa más evidente en base derecha. A partir del día 36 recuperación de la neutropenia con mejoría lenta del cuadro clínico apreciándose en la TC (tras 3 semanas) mejoría de la afectación alveolar y sin apreciarse cavitaciones. Tras recuperación clínica y remisión completa en médula ósea se continúa tratamiento de consolidación según protocolo. Tras 30 días de tratamiento con voriconazol intravenoso se pasa a la presentación oral. Las radiografías posteriores se fueron normalizando suspendiéndose voriconazol tras 260 días (finalizada la última reinducción del matenimiento) tras una TC torácica normal y prueba de detección de galactomanano ().
La evolución posterior ha sido favorable con remisión completa de su enfermedad y asintomática tras 4 meses de suspender tratamiento antifúngico.
Comentarios: En la profilaxis secundaria antifúngica en niños con infección por Aspergillus que precisan continuar con tratamiento quimioterápico, voriconazol oral supone una opción eficaz, como en nuestro caso, aunque son necesarios estudios más amplios para llegar a conclusiones.
Linfohistiocitosis hemofagocítica refractaria. Evolución fatal por infección multisistémica porFusarium
M.I. Hernández, J. López, C. Melero, J.L. Vivanco y M. Baro
Unidad de Hemato-Oncología Pediátrica. Hospital Universitario 12 de Octubre. Madrid. España.
Introducción: La linfohistiocitosis hemofagocítica (LHH) es una rara enfermedad, 1,2 casos/1 millón de niños/año, letal sin tratamiento. Puede ser de origen primario (genética) o secundaria, principalmente debida a infecciones. Las causas más frecuentes de muerte en esta enfermedad son la sepsis por gérmenes oportunistas, la hemorragia y la afectación de sistema nervioso central. Describimos el caso de una paciente con LHH refractaria a tratamiento que fallece por una infección fúngica por Fusarium, un hongo patógeno oportunista emergente y altamente resistente a la mayoría de los antifúngicos y de muy mal pronóstico en pacientes con inmunosupresión grave.
Caso clínico: Niña de 3 años que es ingresada por pancitopenia grave, con cifras de linfocitos normales. En la exploración física destacaba la palidez y la ausencia de adenopatías y organomegalias palpables; fiebre a partir del segundo día de ingreso. En las pruebas complementarias: Hb: 7,1; reticulocitos: 0,7 %; leucocitos: 1.980 (200 neutrófilos); plaquetas: 92.000; ferritina: 2.602, triglicéridos: 269; receptor soluble IL-2 14.000, aspirado y biopsia de médula ósea (MO) con aplasia grado 3/3 con hemofagocitosis; el estudio de perforinas fue normal. Con el diagnóstico de LHH, se efectuaron los estudios sobre la etiología del cuadro que fueron todos negativos. Se inició tratamiento según protocolo SIOP-HHL-2004, difiriendo las primeras 2 semanas el etopósido por la aplasia medular. Durante el tratamiento progresó la pancitopenia, precisando transfusiones de concentrados de plaquetas y hematíes y la neutropenia pasó a ser muy grave. Como complicaciones de la enfermedad presentó infección sistémica por Candida albicans tratada con anfotericina B liposomal e infección por Klebsiella tratada con meropenem ambas con buena evolución; también presentó hemorragia digestiva grave con shock hipovolémico que también pudo resolverse. A los 30 días de tratamiento presenta exantema máculo-eritematoso que evoluciona a pápula y posteriormente a pústula central; aparece también empeoramiento del estado general. En hemocultivos y la biopsia cutánea aparece Fusarium spp. añadiéndose voriconazol. En los días posteriores continúa sin respuesta al tratamiento con nuevo aspirado de MO sin cambios con respecto a previos. Progresivamente se agrava el estado general presentando afectación pulmonar progresiva falleciendo en las siguientes 24 h; tras 47 días de ingreso.
Comentario: La paciente presentada tuvo una evolución fatal debido a la no respuesta a la terapia de su aplasia medular, que propició la infección oportunista por distintos gérmenes. En la LHH la presencia de aplasia no es infrecuente en las fases avanzadas del cuadro, pero en esta paciente llama la atención la precocidad de su presentación y la absoluta falta de respuesta al tratamiento.
La infección por Fusarium es actualmente una infección fúngica emergente en pacientes con trasplante de órgano sólido y en los muy inmunodeprimidos como este caso. A pesar del tratamiento antifúngico combinado la curación de esta enfermedad requiere la recuperación de la función inmunitaria.
Irinotecán y temozolamida en el tratamiento de tumores sólidos refractarios pediátricos
J.A. Blumenfeld Olivares, A. Lassaletta Atienza, A. Pérez Martínez, T. Contra Gómez, C. Scaglione Ríos, N. Martín Ramos y L. Madero López
Servicio de Hemato-Oncología y trasplante hematopoyético. Hospital Universitario Niño Jesús. Madrid.
Introducción: En estudios experimentales con animales se ha comprobado el efecto sinérgico de temozolamida e irinotecán, sin observar un aumento de sus efectos secundarios. En ensayos clínicos posteriores se confirmó la seguridad de la combinación irinotecan + temozolamida en pacientes pediátricos con tumores sólidos refractarios a los tratamientos convencionales.
Objetivo: Valorar la eficacia clínica de la combinación irinotecan + temozolamida en pacientes pediátricos con tumores sólidos metastáticos y refractarios a, al menos dos líneas de tratamiento convencional.
Material y métodos: Cuatro pacientes (3 varones y 1 mujer) con tumores sólidos metastáticos avanzados recibieron un total de 14 ciclos de temozolamida (100 mg/m2/día, días + 1 al + 5) e irinotecán (20 mg/m2/día, días + 1 al + 5 y del + 8 al + 12). Los ciclos se repitieron cada 4 semanas. Se realizó una evaluación completa inicial antes de iniciar el tratamiento y una nueva evaluación al finalizar el tratamiento. Como profilaxis de la diarrea secundaria al irinotecán se administró a todos los pacientes cefixima oral (8 mg/kg/día) desde 3 días antes de iniciar cada ciclo. Las características de los pacientes se muestran en la tabla 2.
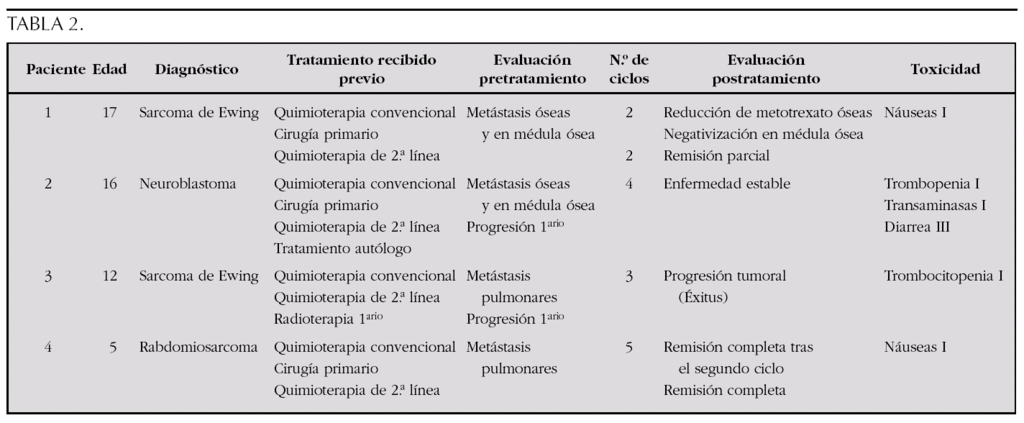
Resultados: Todos los pacientes recibieron los ciclos de forma ambulatoria. La tolerancia del tratamiento fue excelente. No existió mielosupresión moderada o grave con el tratamiento. Tan sólo la paciente número 2 tuvo que ser ingresada para fluidoterapia intravenosa por diarrea moderada-grave tras el tercer ciclo recibido. En ninguno de los casos se suspendió la quimioterapia por toxicidad durante el tratamiento. De los 4 pacientes tratados, uno adquirió la remisión completa de su enfermedad, otro presentó una remisión parcial, otro enfermedad estable y sólo uno progresó a pesar del tratamiento falleciendo 3 meses tras finalizar el último ciclo.
Conclusiones: La combinación temozolamida e irinotecán ha demostrado en estudios previos actividad frente a tumores sólidos pediátricos refractarios a otras líneas de tratamiento. En nuestra serie, la tolerancia ha sido excelente, permitiendo la administración de la quimioterapia de forma ambulatoria, reduciendo los costes y mejorando la calidad asistencial al paciente. A pesar de las limitaciones de nuestra serie en cuanto a número de casos, los resultados en nuestros pacientes son muy esperanzadores de cara al futuro. La combinación temozolamida + irinotecán debe ser analizada en un mayor número de pacientes como tratamiento de rescate en tumores sólidos refractarios a los tratamientos convencionales.
Vinblastina en el tratamiento de gliomas de bajo grado en niños
E. Blanco, R. Silvestre, C. Moscardó, M. Tasso y C. Esquembre
Unidad de Oncología Pediátrica. Hospital General Universitario de Alicante. España.
Introducción: La quimioterapia (QT) es una alternativa en el manejo de los gliomas de bajo grado (GBG) pediátricos no resecables, permitiendo retrasar el uso de radioterapia (RT) en niños de corta edad. No existe consenso en cuanto a drogas y esquemas terapéuticos. La vinblastina (VBL) a dosis de 6 mg/m2 semanal se ha utilizado en niños con GBG que desarrollaron reacciones de hipersensibilidad al carboplatino, con distintos grados de respuesta y sin toxicidad significativa. Estudios preclínicos han demostrado que, administrada a dosis bajas, posee actividad antiangiogénica. Esta característica podría resultar útil en el tratamiento de los GBG, tal y como apuntan algunos estudios descritos en la literatura.
Objetivo: Analizar la eficacia y toxicidad del tratamiento con VBL en niños con GBG.
Matrial y métodos: Estudio retrospectivo de 4 niños con GBG tratados con VBL a dosis de 6 mg/m2 semanal en nuestra Unidad entre 2003 y 2006. En dos casos se administró como tratamiento de segunda línea por ser refractarios y en otros dos como tratamiento inicial.
Resultados: Se describen en la tabla 3:
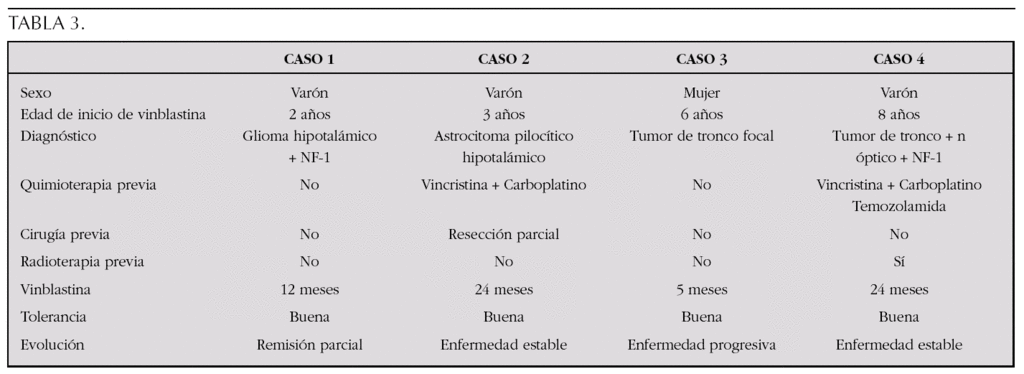
Conclusiones: La utilización de VBL en pacientes con GBG es particularmente atractiva por tratarse de un fármaco eficaz con excelente tolerancia incluso durante períodos prolongados de tiempo. Estas características permiten cumplir objetivos como retrasar la RT en casos seleccionados o mantener estable la enfermedad en casos refractarios, con buena calidad de vida. Es necesario desarrollar estudios clínicos más amplios para definir el papel de dicha droga en el tratamiento de este tipo de tumores pediátricos.
Recidiva tardía de neuroblastoma
F. Lendínez Molinos1, A. Ruiz Sánchez1, G. Cara1, M.A. Vázquez López1 y C. Márquez2
1Unidad de Hemato-Oncología Pediátrica. Servicio de Pediatría. Hospital Torrecárdenas. Almería. 2Unidad de Oncología Pediátrica. Hospital Virgen del Rocío. Sevilla. España.
Introducción: El neuroblastoma (NB) es el tumor abdominal más frecuente de la infancia y típicamente aparece en los primeros años de la vida. Su comportamiento clínico y biológico es peculiar, dado que puede regresar espontáneamente o mostrar una alta agresividad. Las recidivas tardías consideradas como aquellas que se producen más allá de los 5 años del diagnóstico son muy raras. En este sentido, presentamos el caso de un paciente que sufrió una recidiva de NB 16 años más tarde del diagnóstico primitivo. Reflexionamos sobre la incertidumbre pronóstica.
Caso clínico: Paciente varón de 19 años que ingresa para estudio de tumoración laterocervical y axilar izquierda de una semana de evolución, sin signos de infección aparente. La exploración no mostró otros hallazgos patológicos. A los 3 años de vida fue diagnosticado de neuroblastoma abdominal suprarrenal izquierdo estadio 3. Fue remitido a hospital de referencia para tratamiento consiguiéndose la remisión completa y mantenida. La TC cervical realizada al ingreso en nuestro centro mostró un conglomerado adenopático supraclavicular e infraclavicular izquierdo. Con la sospecha de un segundo tumor maligno, la masa fue biopsiada siendo el diagnóstico histológico compatible con neuroblastoma. En la analítica destacó: LDH: 596 U/l; ferritina: 347 ng/ml; enolasa neuronal específica: 85,5 ng/ml; catecolaminas en orina de 24 h elevadas: ácido vanilmandélico 12,6 mg, ácido homovanílico 52,5 mg y dopamina 16.168 μg. El estudio de extensión tumoral incluyó: TC toracoabdominal: masa heterogénea en zona suprarrenal izquierda de 10 × 5 × 5 cm junto a grandes vasos que se extiende retrocruralmente a mediastino posterior. Gammagrafía con MIBG: captación patológica del radioisótopo a nivel suprarrenal izquierdo, laterocervical izquierdo y subclavia izquierda. Biopsia de médula ósea: no evidencia de metástasis. Con el diagnóstico de recidiva local y metastásica de NB se realiza una segunda biopsia de la tumoración cervical para estudio genético, observándose ganancia de N-myc y deleción del 1p. El paciente se incluyó en el protocolo SIOP para neuroblastomas de alto riesgo (HR-NBL-1). Posteriormente ha recibido auto-TPH y tratamiento con cis-retinoico. Tras 5 meses de seguimiento sin tratamiento ha presentado nueva recidiva local en región axilar y abdomen. Actualmente se encuentra en tratamiento con topotecán y temozolamida.
Comentarios: El caso presentado nos hace reflexionar sobre: 1. Cuánto tiempo esperar antes de considerar curado un NB? El curso indolente de la enfermedad ha hecho que algunos autores utilicen el término de NB crónico para referirse a los casos de recidivas tardías. A pesar de su infrecuencia es obvia la necesidad de recomendar un seguimiento prolongado en pacientes con NB. 2. Qué desenlace debe esperarse? La mayoría de los casos provienen de enfermedad primaria avanzada o metastásica y la mortalidad comunicada es alta. A pesar de la agresividad del tratamiento en la segunda recidiva no hemos podido evitar la tercera recidiva, lo que orienta hacia un tumor resistente a la quimioterapia.
Tratamiento con topotecán en niños con neuroblastoma 4 resistente a terapéutica inicial o en recaída
J. Balaguer Guill1, A. Cañete Nieto1, V. Castel Sánchez1, T. Acha2, A. Navajas3 y T. Contra4
1Unidad de Oncología Pediátrica y Fundación para la Investigación La Fe, Hospital La Fe, Valencia. 2Hospital Carlos Haya Málaga. 3Hospital Cruces Vizcaya. 4Hospital Niño Jesús. Madrid. Grupo de Neuroblastoma SEOP. España.
Introducción: El neuroblastoma es uno de los tumores sólidos más frecuentes en la infancia. A pesar de los actuales tratamientos, la supervivencia en estadios avanzados es insatisfactoria, con un porcentaje importante de pacientes que recaen o progresan. El topotecán ha demostrado actividad en el neuroblastoma en ensayos fase II, con una buena farmacocinética, siendo la mielotoxicidad el efecto secundario más importante.
Pacientes y métodos: Estudio prospectivo cuyos criterios de inclusión son: Pacientes mayores de un año diagnosticados de neuroblastoma de cualquier estadio que hayan recaído. Pacientes estadio 4 que sean refractarios al tratamiento inicial de inducción. Pacientes menores de un año que hayan recaído con metástasis óseas o de médula ósea. Todos ellos deben tener un aclaramiento de creatinina > 40 ml/m/1,73 m2, deben haber pasado al menos 14 días desde el último ciclo quimioterápico, es necesario el consentimiento de los padres.
Se administran 2 ciclos de topotecán 1,5 mg/m2/día × 5 días × 2 semanas (cada 3 semanas); seguidos de 2 ciclos de ciclofosfamida + topotecán y 2 de ifosfamida + topotecán.
Resultados: Iniciaron el protocolo 20 pacientes (13 niños y 7 niñas), todos eran mayores de un año. El motivo de inclusión fue: recaída o metástasis (17/20) y no respuesta al tratamiento de inducción en 3 casos. Los protocolos de tratamiento que habían llevado antes de entrar en este estudio fueron: N-I-87 en 6 casos, N-II-92 en 8, N-AR-99 en 5, otros en un caso. El estadio al diagnóstico era: 1 en 1 caso, 2A en 1 caso, 3 en 4, 4 en 14. Presentaban tumoración local 16/20 (en 4 era la única manifestación de la enfermedad), y 17 presentaban metástasis (en médula ósea = 10, óseas = 13, hepáticas = 2, ganglionares = 3, múltiples = 10). La LDH estaba elevada en 5 casos, la ENE en 13 casos. La MIBG fue positiva en 19/20 (en el tumor en 5, en las metástasis en 7, en tumor + metástasis en 7) y negativa en 1. En 7 casos se realizó cirugía al inicio (biopsia = 4, resección = 2, resección con resto macroscópico = 1). Trece pacientes presentaron toxicidad hematológica grado 4 (65 %), 2 pacientes grado 3 (10 %). Diez pacientes presentaron toxicidad infecciosa (50 %), grado 3 en 2 casos, grado 2 en 5 casos, grado 1 en 3 casos. Toxicidad neurológica en 1 caso. Once pacientes tuvieron que ser hospitalizados, recibiendo GCSF 10 de ellos, transfusión de concentrado de hematíes 12, de plaquetas 14 y antibióticos 9. Quince pacientes recibieron al menos 2 ciclos de quimioterapia. La respuesta tras los 2 ciclos fue: RC = 4, RP = 4, Rmixta = 1, PE = 3, fallecieron = 5. Se realizó cirugía durante el quinto ciclo en 9, durante el sexto en 6. Evaluación final: 18 pacientes fallecieron, uno por el tratamiento, otro por neumonía y 16 por progresión de la enfermedad. Dos pacientes siguen vivos (uno con tumor local y otro libre de enfermedad), el primero tenía al diagnóstico enfermedad local + médula ósea y el otro tenía metástasis óseas. La media de tiempo de supervivencia libre de progresión desde la inclusión en el protocolo ha sido de 13,41 meses (rango: 1,43-28,43).
Conclusiones: A pesar de que el neuroblastoma suele ser resistente o con respuesta parcial en pacientes que han recaído o que presentan metástasis, se ha conseguido un buen control de la enfermedad en 2/20 pacientes tratados (uno vive libre de enfermedad), ambos presentaban metástasis al diagnóstico (uno en médula ósea y otro en hueso)¸ el tiempo de seguimiento ha sido de 56 y 42 meses, respectivamente. En los pacientes que han fallecido, se ha conseguido aumentar el tiempo libre de progresión.
Experiencia clínica con una nueva terapia oncolítica antimetastásica
J. García-Castro1, R. Alemany2, M. Ramírez1, A. Lassaletta1, A. Balas3 y A. Pérez-Martínez1, I. González Mediero4, S. Navarro5, J.L. Díaz6 y L Madero1
1Oncohematología y Trasplante. Hospital Niño Jesús. Madrid. 2Instituto Catalán de Oncología. Barcelona. 3Centro de Transfusión. Madrid. 4Anatomía Patológica. Hospital Niño Jesús. 5Hospital Clínico Universitario. Valencia. 6Medicina Nuclear. Hospital Ramón y Cajal. Madrid. España.
Objetivos: Nosotros publicamos recientemente (García-Castro et al. Cancer Gene Ther 2005;12(4):341-9) que las células tumorales autólogas pueden usarse como vehículos para transportar terapias antitumorales a las metástasis. Hemos iniciado un estudio clínico aprobado por el Comité de Ética en Investigación Clínica, para el tratamiento de niños con tumores sólidos metastásicos refractarios.
Material y métodos: El tratamiento consiste en la infusión intravenosa de células tumorales autólogas transfectadas con un adenovirus oncolítico de replicación condicionada (ICOVIR-5). Las células tumorales infundidas alcanzan las metástasis, y el virus destruye las lesiones tumorales con mínima toxicidad.
Resultados: En este trabajo presentamos la experiencia clínica acumulada hasta la fecha:
Caso 1: Varón de 3 años diagnosticado de neuroblastoma estadio IV, con progresión de la enfermedad tras 2 líneas de quimioterapia, y con enfermedad metastásica en pelvis, vértebras y fémures. Se cultivaron células de neuroblastoma obtenidas de aspirados medulares. Estas células se inactivaron mediante irradiación (3.000 rads), se transfectaron con ICOVIR-5 y se infundieron por vía intravenosa. Durante 10 semanas el paciente recibió 4 infusiones de las células manipuladas de esta manera. Se controló la toxicidad mediante estudios clínicos y analíticos. Se estudió la respuesta inmunitaria mediante análisis por citometría de flujo y ensayos funcionales de citotoxicidad. Se estudió la presencia del virus en sangre y orina (PCR), y en muestras obtenidas de aspirados y biopsias medulares (inmunohistoquímica y microscopia electrónica). Se evaluó el resultado del tratamiento mediante gammagrafía con MIBG. El paciente alcanzó la situación de muy buena remisión parcial tras esta terapia, sin toxicidad detectable, y se le indicó un trasplante autólogo.
Caso 2: Varón de 4 años diagnosticado de neuroblastoma estadio IV, que presenta recaída al año de recibir autotrasplante de progenitores de sangre periférica. Afectación ósea en miembros superiores e inferiores, pelvis y columna. Hasta la fecha ha recibido dos infusiones de células, según lo explicado anteriormente. Este paciente será evaluado según lo ya comentado para el caso anterior.
Conclusiones: Los resultados son muy preliminares y han de ser considerados con la debida cautela.
Nuestra estrategia terapéutica parece ser bien tolerada y no se ha asociado con toxicidad aguda ni retardada. El tratamiento mixto (celular y génico) puede resultar beneficioso en niños con tumores sólidos metastásicos refractarios a los protocolos de quimioterapia actuales. Se necesita ampliar la experiencia clínica para valorar el potencial de esta estrategia.
Éxito del tratamiento con rituximab en el síndrome linfoproliferativo postrasplante
M.I. Hernández, M. Baro, J. López, J.L. Vivanco y C. Melero
Unidad de Hemato-Oncología Pediátrica. Hospital Universitario de 12 de Octubre. Madrid. España.
Introducción y objetivos: Con el aumento del número de trasplantes en la edad pediátrica y el desarrollo de nuevos tratamientos inmunosupresores más potentes se está produciendo un incremento paralelo de los procesos linfoproliferativo postrasplante (LPPT). Esta patología es más frecuente en pacientes menores de 5 años, en los seis primeros meses postrasplante. En un 85 % de los casos se asocia al virus de Epstein-Barr (VEB), especialmente en pacientes con serología negativa antes del trasplante e injerto positivo, ambas situaciones frecuentes en pediatría. El objetivo de esta comunicación es describir el éxito del rituximab en el tratamiento de esta patología en dos pacientes.
Pacientes, métodos y resultados
Paciente 1: Varón diagnosticado a los 3 meses de vida de síndrome de Alagille con colestasis intrahepática que es sometido a trasplante hepático ortotópico completo a los 4 años, presentando serologías para virus de Epstein-Barr (VEB) y citomegalovirus (CMV) negativas (injerto VEB desconocido y CMV +). Recibe profilaxis inmunosupresora con tacrolimus y prednisona. A los 4 meses postrasplante presenta serología VEB IgM + y reacción en cadena de la polimerasa (PCR) VEB = 1.500 copias. Dos meses después en ecografía rutinaria se observan varias adenopatías retroperitoneales, por lo que se realiza una TC toracoabdominal donde se visualizan adenopatías mediastínicas, retroperitoneales y varios nódulos pulmonares; en la endoscopia gástrica aparecen varias lesiones necróticas, objetivándose en la biopsia proceso LPPT, linfoma B difuso de células grandes CD20+. Se suspende el tratamiento inmunosupresor, iniciándose tratamiento con aciclovir y rituximab a 375 mg/m2/semana, con un total de 8 dosis, obteniéndose remisión completa que persiste 17 meses después.
Paciente 2: Varón diagnosticado en los primeros meses de vida, de atresia de vías biliares extrahepáticas y colestasis de vías biliares intrahepáticas. Se añade una fibrosis progresiva que precisa trasplante hepático ortotópico completo a los 11 años, siendo VEB + y CMV (injerto CMV + y VEB +). Cinco años después, estando en tratamiento inmunosupresor con tacrolimus es diagnosticado de proceso LPPT Burkitt-like, CD20+, por biopsia de adenopatía preauricular palpable. En el estudio de extensión, se observa además afectación mediastínica. Se inicia tratamiento quimioterápico (en otro centro) completándose sólo 2 ciclos por aparición de rechazo agudo-crónico grave, por lo que es derivado a nuestro centro. Se decide cambiar el tratamiento inmunosupresor a ciclosporina y corticoides a dosis altas, e iniciar tratamiento con rituximab hasta un total de 6 dosis, con lo que se consigue una remisión completa. Posteriormente se pudo realizar un segundo trasplante hepático con éxito, permaneciendo en remisión completa en la actualidad.
Conclusión: En nuestro servicio han sido tratados 5 pacientes por síndrome LPPT monoclonal, siendo los dos últimos, que presentaban anticuerpo CD20+, tratados con rituximab con éxito. En el primer caso el tratamiento fue administrado en monoterapia con una respuesta espectacular. En el segundo caso además esta alternativa es especialmente útil por la grave alteración de la función hepática que impedía el uso de citostáticos. Por ello consideramos este tratamiento, con o sin quimioterapia a bajas dosis, como la mejor alternativa en los procesos LPPT monoclonales por su efectividad y la buena tolerancia observada en ambos pacientes.
Tratamiento con 2-clorodesoxiadenosina en niños con histiocitosis
C. Moscardó, E. Blanco, R. Silvestre, M. Tasso y C. Esquembre
Unidad de Oncología Pediatrica. Hospital General Universitario. Alicante. España.
Introducción: Las terapias que habitualmente se han utilizado en la histiocitosis son efectivas, pero las frecuentes recidivas y resistencias plantean dilemas terapéuticos. En los últimos años se está investigando sobre el uso de la 2-clorodesoxiadenosina (2-CdA) en estos pacientes. Se trata de un análogo de las purinas con un doble efecto, antiproliferativo e inmunomodulador. No hay acuerdo en la dosificación y forma de administración. Se describen ciclos de 3 a 13 mg/m2/día durante 3 o 5 días, en perfusión de entre 2-24 h, con intervalos de 21-28 días y un máximo de 6 ciclos.
Objetivo: Analizar la eficacia, seguridad y forma de administración de la 2-CdA en niños con histiocitosis recurrente o refractaria.
Material y métodos: Estudio retrospectivo de 6 niños, cinco con histiocitosis de células de Langerhans (2 recurrentes y 3 refractarias) y uno con enfermedad de Rosai-Dorfman refractaria, tratados con 2-CdA en nuestra unidad entre 1999 y 2005. Se describen los siguientes datos de todos los pacientes: edad, sexo, órganos afectados y tratamientos previos; dosis, forma de administración, respuesta y toxicidad con la 2-CdA.
Resultados: Los pacientes fueron en su mayoría varones (5:1), con una edad mediana de 2,3 años (9 meses-8,5 años). Todos presentaban enfermedad multisistémica, uno de ellos con afectación de órganos de riesgo. La 2-CdA fue tratamiento de segunda línea en cuatro de los casos y de tercera línea en dos. En 4 niños se administraron dosis bajas (5-6,5 mg/m2/día × 3 días, 6 ciclos), en uno intermedias (7 mg/m2/día × 5 días, 2 ciclos) y altas (14 mg/m2/día × 3 días, 4 ciclos) en una paciente por falta de respuesta a dosis bajas. De los 31 ciclos administrados, 18 fueron mediante infusión continua y 13 en infusión de 3 h. La paciente tratada con altas dosis obtuvo muy buena respuesta parcial pero presentó toxicidad hematológica de grado IV con afectación de las 3 series, lo que obligó a suspender el tratamiento. El cuadro se resolvió progresivamente en 6 meses sin haber presentado complicaciones graves durante el período de aplasia. A dosis bajas e intermedias no se observaron toxicidades significativas. Tras una mediana de seguimiento de 3,5 años, 4 pacientes se hallan en remisión completa (3,5-8,6 años), uno en muy buena respuesta parcial (9 meses) y uno falleció por progresión de la enfermedad.
Discusión: En nuestra experiencia la 2-CdA se ha mostrado eficaz en histiocitosis recurrentes/refractarias. La toxicidad es sobre todo hematológica y directamente relacionada con la dosis. A dosis bajas e intermedias es muy bien tolerada, mientras que a dosis altas pueden llegar a producir aplasia medular prolongada en el tiempo, aunque no se han descrito hasta el momento complicaciones graves.
Tal y como aparece en la literatura especializada, la infusión en 3 h frente a la continua parece igual de eficaz sin aumentar la toxicidad, y posibilita su administración de forma ambulatoria.
El perfil de eficacia y tolerancia de la 2-CdA permite su utilización precoz en histiocitosis de evolución tórpida. Se necesitan más estudios para determinar la dosis, formas de administración e indicaciones óptimas para cada paciente.
Fracaso de nuevos tratamientos quimioterápicos
N. Nieto, M. García Arias, M. Guibelalde y J.A. Salinas
Unidad de Hemato-Oncología Infantil. Hospital Son Dureta. Palma de Mallorca. España.
Introducción: Las terapias antineoplásicas novedosas suponen una esperanza en el tratamiento de tumores de alto riesgo, pero su papel está todavía por determinar. Presentamos 2 casos clínicos, como ejemplo del fracaso de 2 terapias: el irinotecán (inhibidor específico de la ADN-topoisomerasa-I) y el gentuzumab (anticuerpo monoclonal anti-CD33).
Caso clínico 1: Paciente de 5 años, diagnosticado a los 4,5 años de hepatoblastoma no metastático estadio Pretext-4. Recibió tratamiento según protocolo SIOPEL 3. Posteriormente se realizó un trasplante hepático ortotópico parcial de donante cadáver. Alcanzó la remisión completa. Dos meses después el paciente desarrolló un linfoma no hodgkiniano plasmoblástico, que fue tratado según protocolo BFM-95 para LNH B rama 2, alcanzando igualmente la remisión completa. En una TC realizada un mes después se detectaron varias masas pulmonares: 3 nódulos en lóbulo superior derecho y uno en el inferior izquierdo, coincidiendo con una elevación de la a -fetoproteína sérica (AFP). Ante la sospecha de metástasis pulmonares de hepatoblastoma se inició tratamiento con irinotecán intravenoso. En nuestro paciente se administraron 4 ciclos consecutivos de irinotecán, 20 mg/m2/dosis (10 días/ciclo). La tolerancia al tratamiento fue buena. La reevaluación tras la administración del último ciclo mostró dos nuevas imágenes pulmonares y una marcada elevación de AFP: se suspendió el medicamento por progresión tumoral. Posteriormente se administraron 2 ciclos de 5-fluorouracilo de 600 mg/m2 y 4 ciclos de docetaxel a 100 mg/m2 y se realizó en dos tiempos la exéresis quirúrgica de todas las metástasis pulmonares. A pesar de todo, la AFP continúa elevándose y queda pendiente nueva reevaluación terapéutica.
Caso clínico 2: Paciente de 13 años diagnosticado de leucemia mieloide aguda FAB M1 de alto riesgo, en junio de 2004 [> 100.000 leucocitos y genética 46XY/46XY t(4,12); del(12)(p13)]. Inició tratamiento según protocolo SHOP-LANL 2001, consiguiendo remisión completa. Se consolidó con autotrasplante de progenitores hematopoyéticos (PH) (busulfán, ciclofosfamida y VP 16). Tres meses después recidivó (80 % de blastos con fenotipo similar al diagnóstico y nódulo pulmonar), tratándose con quimioterapia FLAG-IDA. Se inicio búsqueda de donante no emparentado de PH. Aunque alcanzó remisión tras el primer ciclo, volvió a recaer 6 meses después, por lo que se decidió tratarle con gentuzumab (9 mg/m2) × 2. Tras la primera dosis desarrolla adenopatía submandibular derecha (células mieloides en PAAF). Se decide administrar la segunda dosis de gentuzumab y radioterapia cervical, a pesar de lo cual la enfermedad progresa (adenopatías cervicales y masa tímica con efusión pleural). Dada la refractariedad del caso, se inician cuidados paliativos, falleciendo el paciente a los 2 meses de la segunda recidiva.
Conclusiones: 1. Las nuevas terapias no siempre ofrecen beneficios a los pacientes oncológicos. 2. Se desconoce en qué momento de la enfermedad deberían ser utilizados estos fármacos. 3. Son necesarios estudios más amplios que evalúen su eficacia en patología oncológica.
