



Murcia, junio de 2004
Proteína C reactiva ultrasensible: ¿Marcador precoz de riesgo de hipertensión en hijos de hipertensos esenciales?
S. Málaga 1, J.J. Díaz 1, I. Málaga 1, J. Argüelles 2, M.ª A. Diéguez 3 y M. Vijande 2
1Áreas de Pediatría y 2Fisiología. Facultad de Medicina. Universidad de Oviedo. 3Servicio de Inmunología. Hospital Central de Asturias. Oviedo. España.
Introducción: Los métodos tradicionales de determinación de proteína C reactiva (PCR) no son suficientemente sensibles para detectar los niveles que presentan los individuos sanos. En la actualidad se han desarrollado métodos de detección de PCR ultrasensible aplicables en población sana, validados internacionalmente. Diversos estudios en adultos han demostrado una relación positiva de la PCR con la obesidad, presión arterial (PA) y sensibilidad a la insulina. Los escasos estudios realizados en niños y adolescentes han relacionando principalmente el sobrepeso y la PA sistólica (PAS) con los valores de PCR.
Objetivo: Determinar el comportamiento de la PCR, así como su relación con la distribución de la PA y los factores de riesgo cardiovascular (FRCV) clásicos, en una población de niños y jóvenes sanos.
Se trata de un estudio transversal de casos y controles.
Sujetos de estudio: Grupo índice: 51 niños y jóvenes sanos (28 varones), edad media 16,9 ± 4,7 años, con al menos un progenitor portador de HTA esencial. Grupo control: 73 niños y jóvenes sanos (43 varones), edad media 16,1 ± 2,4 años, procedentes del estudio longitudinal RICARDIN, sin antecedentes de HTA en progenitores. Determinaciones: parámetros antropométricos, PA (mmHg) media de dos mediciones y en suero: colesterol total, HDL-colesterol, LDL-colesterol, triglicéridos (mg/dl) y PCR (mg/l). El programa estadístico utilizado fue el SPSS versión 11.0 para el entorno Windows. El estudio fue financiado con las siguientes ayudas de investigación: Fundación Ernesto Sánchez Villares-2003; Universidad de Oviedo MA-03-519-1 e Instituto de Salud Carlos III 03/0350.
Resultados: No se observaron diferencias significativas entre ambos grupos en cuanto a sexo, edad, peso, talla, índice de masa corporal (IMC) y cifras tensionales: PAS (mmHg) controles: 111,95 ± 10,7 frente a hijos HTA 112,67 ± 12,11. PA diastólica controles: 70,05 ± 8,12 vs hijos HTA 68,85 ± 7,95). Tampoco se hallaron diferencias en los valores del colesterol total, HDL-colesterol y LDL-colesterol. Sí se encontraron diferencias en las cifras de triglicéridos (controles: 52,85 ± 23,07; hijos HTA: 71,92 ± 44,43; p = 0,006) y PCR (controles: 0,77 ± 2,05; hijos HTA: 1,14 ± 1,56; p = 0,001). Igualmente se observó una clara correlación entre los valores de PCR y el IMC en ambos grupos.
Conclusión: Los valores de PCR en hijos de enfermos con HTA esencial se mostraron significativamente más altos que los del grupo control, lo que sugiere que la PCR pudiera comportarse como un FRCV precoz e importante para el desarrollo de HTA esencial.
Presión arterial y obesidad en hijos de hipertensos esenciales: independencia del eje renina-angiotensina-aldosterona y de la sensibilidad gustativa a la sal
S. Málaga 1, J.J. Díaz 1, I. Málaga 1, J. Argüelles 2, C. Perillán 2, F. Díaz 2, S. Braga 3 y M. Vijande 2
1Áreas de Pediatría y 2Fisiología. Facultad de Medicina. Universidad de Oviedo. 3Servicio de Bioquímica. Hospital Universitario Central de Asturias. Oviedo. España.
Introducción: El aumento de los casos de obesidad está alcanzando cifras epidémicas en los países industrializados. Es conocida la asociación de esta enfermedad con la elevación de los valores de presión arterial (PA) en niños y adolescentes. Las relaciones entre esos dos factores patogénicos, así como su dependencia genética y comportamental son insuficientemente conocidos.
Objetivo: Estudiar la prevalencia de obesidad en hijos de enfermos con hipertensión arterial esencial (HTE) y analizar su relación con la PA, sensibilidad gustativa a la sal y variables de función renal.
Pacientes y métodos: Hijos de HTE: 51 niños y jóvenes sanos (28 varones) de 16,9 ± 4,7 años, con al menos uno de sus progenitores con HTE. Grupo control: 73 jóvenes sanos (43 varones) con una edad media de 16,1 ± 2,4 años, que habían completado el seguimiento longitudinal del estudio RICARDIN. Se consideraron obesos los participantes menores de 14 años, con IMC superior al P95 para su edad, y superior a 25 kg/m 2 en el resto. PA: media de dos determinaciones, realizadas con el sujeto sentado y en reposo durante 5 min mediante esfigmomanómetro de mercurio. Test de sensibilidad gustativa salina, para determinar la concentración salina mínima detectable individualmente. En el grupo de hijos de HTE se realizaron además: determinaciones en sangre y orina (micción aislada) de creatinina e iones, para cálculo de función renal estimada (FRE) por la talla, EFNa y EFK, y en plasma, aldosterona, actividad de renina (ARP) y enzima conversora de la angiotensina (ECA), por radioinmunoanálisis. Procedimiento estadístico: descriptivo convencional, comparación de medias (t de Student) y de proporciones (x 2). Financiación: proyecto FIS 03/0350 y Fundación Ernesto Sánchez Villares 2002.
Resultados: En hijos de HTE, la prevalencia de obesidad fue del 33 % frente al 11 % en controles (p = 0,002). No existieron diferencias significativas entre casos y controles para los valores de PA sistólica (PAS) y diastólica (PAD). En los hijos de HTE, el grupo de obesos presentó valores de PAS 10 mmHg más altos que los no obesos (p = 0,004), no existiendo diferencias en los valores medios de PAD. No se observaron diferencias significativas en los valores de FRE, EFNa, EFK, aldosterona, ARP ni ECA. El umbral de sensibilidad gustativa a la sal fue similar en ambos grupos.
Conclusión: Los hijos de sujetos con HTE presentan una alta prevalencia de obesidad en comparación con controles. Los hijos obesos de enfermos con HTE presentan valores de PAS significativamente más altos que los no obesos; estas diferencias no son atribuibles a diferente actividad del eje renina-angiotensina entre ambos grupos, ni a diferente sensibilidad gustativa a la sal.
Factores de hipertensión arterial en el seguimiento de niños trasplantados renales
A. Alonso Melgar, C. Fernández, C. García Meseguer, M. Melgosa, L. Espinosa, A. Peña, J. Bravo y M. Navarro
Servicio de Nefrología Pediátrica. Hospital La Paz. Madrid. España.
Objetivos: Conocer la prevalencia de hipertensión arterial (HTA) en la población trasplantada de nuestro centro así como sus factores determinantes.
Pacientes y métodos: Estudiamos a todos aquellos pacientes trasplantados con visita programada o a demanda durante una semana. Se investigó la presencia actual o previa de HTA, repercusión cardiológica; tipo de donante; episodios de rechazo; inmunosupresión; función renal; grado de control de la HTA y número y tipo de hipotensores utilizados.
Resultados: De los 30 trasplantados estudiados (22 varones y 8 niñas de 14,4 ± 4,8 años de edad y evolución de 4,5 ± 4 años), el 46,7 % presentaron HTA y un 90 % la había presentado en algún momento anterior del trasplante. La población hipertensa estaba bien controlada: 125 ± 10 mmHg de presión arterial sistólica (PAS) (percentil 74 ± 31) y 69 ± 7 de presión arterial diastólica (PAD) (percentil 56 ± 27). Aunque el porcentaje de hipertensos fue similar al de la fase de diálisis, existió una mejoría ecocardiográfica significativa de la hipertrofia ventricular izquierda, desapareciendo en el 30 % de los que la presentaban. Los hipotensores más utilizados fueron los antagonistas del calcio (85,7 %) y los ARA 2 (28 %); un 57 % precisó más de un hipotensor. Los factores que se asociaron con la presencia de HTA fueron: donante cadáver frente a donante vivo; edad inferior del donante; mayor incidencia de episodios de rechazo; niveles mayores de triglicéridos: 101 ± 50 mg/dl frente a 60 ± 24 mg/dl; mayor dosis de esteroides y peores datos de función renal compatibles con nefropatía crónica del injerto: creatinina 1,8 ± 1,2 mg/dl frente a 1,1 ± 0,57 mg/dl; FGE 66 ± 35 ml/min/ 1,73 m 2 frente a 79 ± 29 ml/min/1,73 m 2; proteinuria 0,99 ± 0,9 g/24 h frente a 0,2 ± 0,23 g/24 h; albuminuria: 366 ± 477 μg/ min frente a 38 ± 84 μg/min y cistatina C 2 ± 1 mg/l frente a 1,3 ± 0,5 mg/l.
Conclusiones: La HTA es un problema frecuente de los niños con trasplante renal aunque existe un buen control de la misma. La disfunción crónica del injerto es la causa más importante.
Hipomagnesemia-hipercalciuria con nefrocalcinosis y anomalías oculares
C. Loris 1, M. Justa 1, S. Abio 1, C. Martín 1 y C. Ferrer 2
1Unidad Nefrología Pediátrica. 2Servicio de Oftalmología. Hospital Universitario Miguel Servet. Zaragoza. España.
Objetivo: La hipomagnesemia-hipercalciuria con nefrocalcinosis es una tubulopatía poco frecuente que suele conducir a insuficiencia renal terminal (IRT). Caracterizada por presentar hipomagnesemia-hipercalciuria, nefrocalcinosis, poliuria y polidipsia, puede asociar en grado variable anomalías oculares. Está causada por una mutación del gen PCLN1, el cual codifica a la proteína paracellina-1, implicada en la reabsorción de calcio y magnesio en el segmento grueso de la rama ascendente del asa de Henle. El objetivo de esta comunicación es revisar la evolución en nuestros pacientes y la incidencia de anomalías oculares en nuestros casos y en los casos descritos en España, comparándolos con grupos de otros países.
Material y métodos: Estudio retrospectivo de un grupo de pacientes diagnosticados en nuestro hospital. Revisión de casos publicados en España y resto de países.
Resultados: Se presentan 6 mujeres y 3 varones con poliuria, polidipsia, además de infección urinaria y litiasis. Todos tenían hipomagnesemia-hipercalciuria con nefrocalcinosis. Al diagnóstico cinco presentaban insuficiencia renal; cuatro fueron trasplantados sin recidiva posterior. En 8 (88 %) se encontraron anomalías oculares diversas. El 81 % de los pacientes españoles presentaban anomalías oculares frente al 24 % de otros países.
Conclusión: La evolución a IRT en la segunda-tercera década de la vida es un hecho prácticamente constante. Únicamente los casos diagnosticados en edades tempranas presentan un filtrado glomerular relativamente normal. Las anomalías oculares se perfilan como la manifestación extrarrenal más frecuente en los pacientes españoles (81 %).
El trasplante renal es el único tratamiento que corrige por completo este trastorno tubular sin que haya recaída de la enfermedad.
Hiperoxaluria primaria tipo 1. Una enfermedad conformacional
V.M. García Nieto 1, A. Santana 2, M.I. Luis Yanes 1, M.D. Rodrigo Jiménez 3, C. Marrero Pérez 1 y A. Torres 2
1Nefrología Pediátrica. Hospital Nuestra Señora de la Candelaria. Santa Cruz de Tenerife. 2Hospital Universitario de Canarias. Santa Cruz de Tenerife. 3Hospital Son Dureta. Palma de Mallorca. España.
Introducción: La hiperoxaluria primaria tipo 1 (HP1) es una rara enfermedad de herencia autosómica recesiva, causada por mutaciones en el gen alanina: glioxilato aminotransferasa (AGTX) que codifica una enzima que es importante en la metabolización del glioxilato. Una actividad insuficiente AGTX conduce a un incremento en la conversión de glioxilato en oxalato.
Pacientes y métodos: La paciente 1 fue estudiada a los 9 meses de edad tras observarse 4 cálculos en una radiografía simple de abdomen. A los 22 meses padeció insuficiencia renal aguda por enclavamiento bilateral de los cálculos. El paciente 2, hermano de la anterior, a los 6 años era portador de 3 cálculos ecográficos. La oxaluria de ambos hermanos era de 110,8 y 185,1 mg/día/1,73 m 2, respectivamente. Los niveles de los ácidos glicólico y L-glicérico fueron normales. El paciente 3 fue remitido a los 6 años de edad por haber expulsado 5 cálculos. En la ecografía se observó, además, un cálculo coraliforme bilateral. La oxaluria era de 315,2 mg/día/ 1,73 m 2.
Resultados: Los 3 pacientes son portadores de una mutación en el gen AGTX consistente en un cambio T/C en el nucleótido 853 (exón 7), que corresponde a un cambio Ile a Thr en el residuo 244 de la proteína AGTX (I244T). Aunque I244 no afecta la actividad AGTX o su localización subcelular, cuando está presente en la misma molécula el polimorfismo común Leu-11 a Pro (L11P) se produce una modificación intragénica que da lugar a una proteína mal plegada que tiende a formar agregados inactivos con una menor actividad enzimática en los extractos celulares solubles.
Conclusiones: La mutación I244T en combinación con el polimorfismo L11P en el gen AGTX demuestra que la HP1 es una enfermedad conformacional que podría ser susceptible de tratamiento con chaperones farmacológicos. Su demostración ha confirmado el diagnóstico de HP1, a pesar existir niveles normales de ácido glicólico en dos de los pacientes.
Hiperoxaluria primaria tipo I: heterogenicidad y posibilidades terapéuticas
I. Elorza, M. Tobeña, F. Paredes, R. Vilalta, J. Nieto, F. Castelló y A. Vila
Servicio de Nefrología Pediátrica. Hospital Vall d'Hebron. Barcelona. España.
Objetivo: Describir la heterogenicidad de la hiperoxaluria primaria tipo I y el avance de los conocimientos a su respecto, mediante las conclusiones obtenidas del estudio comparativo de 5 pacientes afectados (tabla 1).
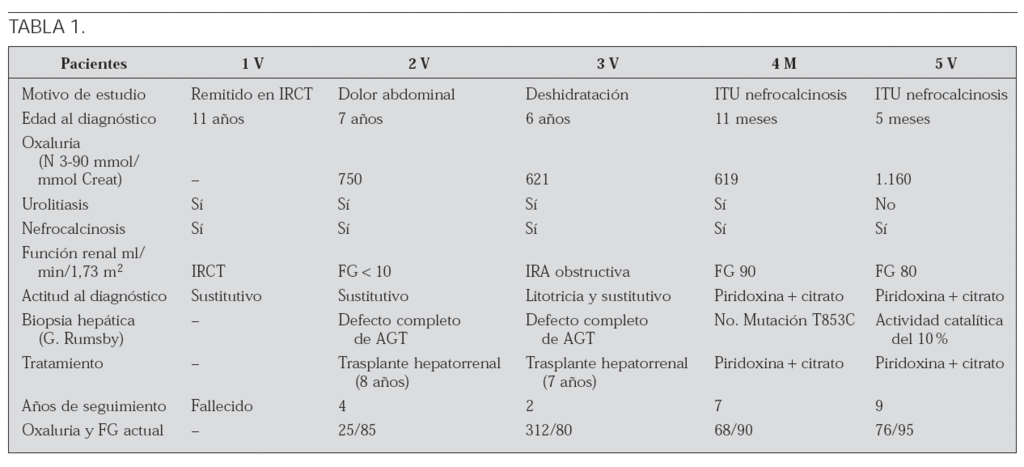
Conclusiones: En los pacientes diagnosticados precozmente, el tratamiento con piridoxina y citrato puede disminuir la síntesis de oxalato y su insolubilidad. El trasplante hepático exclusivo se reserva para los no respondedores, realizándose hepatorrenal si por diagnóstico tardío se encuentran en insuficiencia renal.
Fenotipo de los pacientes cistinúricos detectados por cribado metabólico neonatal en nuestra comunidad
C. Vicente 1, A. Vicente 1, S. Gracia 1, E. Guillén 2, C. González 3 y A. Fernández 3
1Unidad de Nefrología Pediátrica. 2Unidad de Genética Clínica Pediátrica. 3Centro de Bioquímica y Genética Clínica. Hospital Universitario Virgen de la Arrixaca. Murcia. España.
Introducción: La cistinuria es una enfermedad hereditaria, debida a un defecto en el transporte tubular renal e intestinal de cistina y aminoácidos dibásicos. El objetivo del estudio es analizar el fenotipo de los pacientes con cistinuria detectados por cribado neonatal y la presencia de litiasis.
Material y métodos: Se estudiaron 27 pacientes cistinúricos detectados por cribado neonatal en orina y confirmados a los 12 meses entre 1989 y 2001. Se recoge orina de 24 h de los padres y de los pacientes para determinar el fenotipo. Se analiza la incidencia, características fenotípicas y presencia de nefrolitiasis. Se clasifican en homocigotos y heterocigotos según la excreción de cistina (> 250 mg/g creatinina) y tipo I/tipo no I según la excreción de cistina y aminoácidos dibásicos de los padres.
Resultados: Se realizó cribado neonatal a 170.882 recién nacidos (RN), estimándose una incidencia de 1/4.270 RN. Veintisiete pacientes (13 M/14 V). Tipo I homocigoto (HMI) 9, tipo no I homocigoto (HMNI) 4 y 14 tipo no I heterocigoto (HTNI). Cuatro pacientes presentaron litiasis: 2 HMI y 2 HMNI. Edad media de aparición de litiasis: 3 años y 3 meses; sólo un paciente presentó síntomas mientras que en los otros tres la litiasis se detectó de forma casual en ecografía de control. Todos recibieron tratamiento con hiperhidratación y/o alcalinización y sólo uno precisó tratamiento quirúrgico. Se realizó estudio molecular en 9 pacientes de los cuales se detectó mutación en el gen SLC3A1 en dos de ellos, clasificados fenotípicamente como HMI, quedando pendiente el resultado en los otros siete.
Conclusiones: La cistinuria es una causa poco frecuente de litiasis, aunque se debe tener en cuenta como posible etiología de litiasis en la infancia. Una correcta identificación fenotípica y genotípica permitirá una mejor profilaxis y tratamiento de esta enfermedad. El cribado metabólico neonatal en orina permite la identificación precoz de esta patología y posible prevención de litiasis. La incidencia en nuestro estudio es más elevada, probablemente debido a la detección precoz.
Caracterización de nuevas mutaciones del genCLCN5asociadas con la enfermedad de Dent
E. Ramos-Trujillo 1, C. Flores 1, H. González-Acosta 1, L. Espinosa 3, C. Loris 4, R. Oliveros 5, M. Aguirre 5, V. García Nieto 1,2 y F. Claverie-Martín 1
Unidades de 1Investigación y 2Nefrología Pediátrica. Hospital Universitario Nuestra Señora de la Candelaria. Santa Cruz de Santa Cruz de Tenerife. Servicios de Nefrología Pediátrica. Hospital Universitario 3La Paz (Madrid), 4Miguel Servet (Zaragoza) y 5Cruces (Baracaldo). España.
La enfermedad de Dent es una tubulopatía renal caracterizada por proteinuria de bajo peso molecular, hipercalciuria, nefrocalcinosis, nefrolitiasis, raquitismo e insuficiencia renal. En algunos pacientes también se observan otros defectos del túbulo proximal como hiperaminoaciduria, hiperfosfaturia, glucosuria y defecto en la capacidad de acidificación urinaria. Deficiencias funcionales en el gen CLCN5, localizado en el cromosoma Xp11.22, causan la enfermedad. Este gen codifica el canal de cloro ClC-5 que se expresa principalmente en el riñón. Nuestro objetivo es la identificación de mutaciones en CLCN5 de pacientes españoles diagnosticados con la enfermedad de Dent. Hemos analizado, mediante secuenciación directa de los exones codificantes y sus regiones intrónicas flanqueantes, el gen CLCN5 de 3 pacientes que presentaron b2-microglobulina elevada en orina e hipercalciuria. Uno de ellos también mostró nefrocalcinosis y osteoporosis. Se identificaron tres nuevas mutaciones: una transición C a T en el exón 9 que predice el cambio de la arginina 467 por un codón de parada; una transición A a G en el sitio aceptor de splicing del intrón 2 que supone una alteración en el procesamiento del ARN mensajero (ARNm); y una transición C a T en la posición 30 del intrón 9 de efecto desconocido. Este último cambio está localizado en una posible secuencia branchpoint, secuencias poco conservadas que participan en la unión de la maquinaria del splicing. El análisis del gen CLCN5 de 78 cromosomas de controles sanos apunta a que este cambio es una mutación y no un polimorfismo. Sin embargo, tras confirmar la equivalencia entre los ARNm de sangre total y de riñón, el examen del ARNm de la sangre del paciente reveló que esta mutación no causa alteraciones en el procesamiento del ARNm. Estos resultados y otros obtenidos previamente por nuestro grupo indican una heterogeneidad genética de la enfermedad de Dent en España.
Proyecto financiado por la Fundación Canaria de Investigación y Salud FUNCIS.
Litiasis renal: revisión de 5 años
C. Gutiérrez Regidor, C. López-Menchero Oliva, A. de la Huerga López, A. Fernández Escribano, C. Aparicio y E. Izquierdo
Hospital General Universitario Gregorio Marañón. Madrid. España.
Introducción: Revisamos la litiasis renal en los niños de nuestro medio, factores predisponentes, evolución y tratamiento.
Material y métodos: Se analizan retrospectivamente las litiasis seguidas en nuestro servicio, factores asociados y antecedentes familiares.
Resultados: Se estudiaron 20 pacientes de 5 meses a 15 años, 67 % varones. El 68 % tenían antecedentes familiares. Cuatro recibieron diuréticos en el período neonatal y tres de ellos nutrición parenteral. Un paciente asociaba cálculos en parótida. El 60 % refirieron elevada ingesta de lácteos. Motivo de estudio: 35 % microhematuria, 17 % infecciones del tracto urinario, 17 % dolor abdominal recurrente, 11 % dolor abdominal y microhematuria, 6 % hipercistinuria, hallazgo casual 11 %. Hallazgos ecográficos: 24 % litiasis pélvica bilateral, 64 % unilateral, 6 % ureteral y 6 % uretral. En un 12 % existía dilatación pielocalicial unilateral, en un 6 % bilateral, en un 6 % ureterohidronefrosis y nefrocalcinosis; un 12 % asociaba reflujo vesicoureteral. Alteraciones metabólicas: 10 % hipercalciuria, 10 % hiperuricosuria, 20 % asociaba hipercalciuria e hiperuricosuria, 5 % hipercalciuria e hiperoxaluria, 5 % hipercalciuria con hiperoxaluria e hipocitraturia, 5 % hipercalciuria con hiperuricosuria e hipocitraturia, 10 % hiperoxaluria con hiperuricosuria e hipocitraturia, 5 % hipercalciuria con hipocitraturia. El 25 % presentaban hipocitraturia. En el 24 % se objetivó natriuresis elevada (7,5 mEq/kg/día), en el 75 % de los casos asociada a hipercalciuria y en el 25 % a hiperuricosuria. Con el cambio en los hábitos dietéticos la hipercalciuria se corrigió en el 44 %; el 64 % precisó tratamiento farmacológico. El 18 % precisó litotomía y el 15 % litotricia.
Conclusiones: 1. La litiasis renal predomina en varones y en el 70 % de los casos es familiar. 2. La mayoría de los pacientes tenían varios factores predisponentes asociados. 3. En el 50 % el descubrimiento fue en pacientes asintomáticos. 4. La localización más frecuente fue en pelvis. 5. El 65 % tenían alteraciones metabólicas. 6. La mayoría de nuestros pacientes evolucionan favorablemente con actitud expectante.
Litiasis renal pediátrica: experiencia propia
M. Fernández, V. Martínez, R. Pardo, T. Pérez, F.A. Ordóñez, F. Santos y S. Málaga
Sección de Nefrología Pediátrica. Hospital Universitario Central de Asturias. Universidad de Oviedo. España.
Objetivo: Analizar la casuística de litiasis renal pediátrica durante un período de 20 años en un hospital de referencia.
Pacientes y métodos: Estudio retrospectivo de los casos de urolitiasis diagnosticados en los últimos 20 años en nuestro Hospital, analizando sus principales características clínicas analítico-radiológicas y su relación con los factores predisponentes identificados.
Resultados: Durante el tiempo de estudio se diagnosticaron 37 casos de urolitiasis infantil (2 casos/millón de habitantes/año), con una edad media de 5 años y 6 meses (rango: RN a 13 años-11 meses) y una relación V:M de 1,8. Antecedentes familiares de litiasis (en primer y segundo grado) se recogieron en 17 casos (46 %), en tres de ellos de naturaleza metabólica y en dos de enfermedad nefrourológica. La manifestación clínica más frecuente fue el dolor, exclusivo (11 casos) o asociado a hematuria (12); en 6 pacientes se trató de un hallazgo casual. Los factores predisponentes identificados fueron: alteraciones urológicas en 18 niños (49 %), hipercalciuria en 11 casos (30 %), cistinuria en 2 (5 %), infección del tracto urinario (ITU) de repetición en 2 (5 %) y combinación de alguno de los anteriores en 4 (10 %). Urocultivo positivo en el momento del diagnóstico en 11 (29 %); además, en el 73 % de estos niños con ITU coexistía otro factor de riesgo litogénico. En 4 pacientes (10 %) no se constató la presencia factores de riesgo reconocidos. La confirmación de la litiasis se realizó mediante ecografía en 15 casos, ecografía más radiografía simple de abdomen en otros 12; en cuatro mediante radiografía simple y en seis mediante urografía intravenosa. La localización fue exclusivamente renal en 21 niños, ureteral en ocho, vesical en 5 y 9 de los niños presentaban cálculos en diferentes niveles de trayecto urinario. Sólo en 16 de los casos se logró analizar la composición del cálculo, estando en 12 de ellos formado por sales cálcicas. En un niño la litiasis se asoció a IRC con hipocrecimiento.
Conclusiones: 1. La incidencia de urolitiasis en nuestro medio es similar a otras series europeas. 2. Cerca del 50 % de los pacientes presentan antecedentes familiares de urolitiasis, así como uno o más factores predisponentes.
Urolitiasis infantil. Revisión clínica y epidemiológica de los últimos años en nuestro medio
I. Santos Ruiz, E. Hidalgo-Barquero del Rosal, J.M. García Blanco, C. Molina Molina y E. Fernández Calderón
Hospital Materno-Infantil. Complejo Hospitalario Infanta Cristina. Badajoz. España.
Objetivos: Determinar la incidencia y principales características de la urolitiasis infantil en nuestra área de salud, en los últimos años.
Método: Estudio retrospectivo de 58 casos de pacientes diagnosticados y seguidos en la Unidad de Nefrología Infantil en el período comprendido entre 1991 y 2003. Se recogieron y analizaron datos relativos a edad en el momento del diagnóstico, sexo, antecedentes familiares, antecedentes personales relacionados con la formación de urolitiasis (ITU de repetición, anomalías genitourinarias, metabolopatías previas, inmovilización, etc.); forma de presentación; datos de laboratorio (hematología, bioquímica, estudio metabólico y de función renal, etc.); pruebas de imagen; tipo de litiasis (localización, litoquímica, etc.) y datos relacionados con el tratamiento y sus complicaciones, evolución posterior y aparición de secuelas.
Resultados: La prevalencia fue de 1/51 pacientes nuevos atendidos en la consulta de nefrología infantil en este período. La media de edad al diagnóstico fue de 7,2 años, con claro predominio en varones (62,3 %). Presentaban antecedentes familiares de litiasis el 41,5 %. La etiología más frecuente fue la metabólica (65 %) y dentro de esta la hipercalciuria; la litiasis se consideró idiopática en el 24 % de los casos. La forma típica de presentación fue el dolor abdominal asociado o no a hematuria. Dentro de las pruebas diagnósticas la más útil para detectar litiasis fue la ecografía abdominal. En el 44 % se asociaba a repercusión sobre vías urinarias (hidronefrosis, ectasia). La localización más frecuente fue pelvis renal, con predominio en el sistema excretor derecho. A pesar de que el tratamiento médico y la resolución espontánea fueron lo más frecuente, en el 29 % de los casos fue necesario el tratamiento quirúrgico y en el 10 % se empleó litotricia con bastante eficacia. Se observaron recidivas en 13 casos (22,4 %). Las complicaciones del tratamiento fueron infrecuentes. No se han detectado graves secuelas en el seguimiento posterior.
Comentarios: Los resultados obtenidos en nuestro estudio (edad, distribución por sexos, alta incidencia de antecedentes familiares de litiasis y la etiología metabólica predominante), se ajustan a los obtenidos en otras series recientes realizadas en nuestro medio. Destacar la importancia de realizar un estudio metabólico detallado en los pacientes diagnosticados de litiasis que permita identificar la etiología de la misma, dado el aumento de la incidencia de este tipo de litiasis con respecto a la litiasis infecciosa en nuestro medio. La ecografía es la prueba diagnóstica más eficaz en su detección y seguimiento. El tratamiento quirúrgico de la urolitiasis infantil ha experimentado en los últimos años un gran avance hacia terapias cada vez menos invasivas, gracias al desarrollo y perfeccionamiento de técnicas y de instrumentales adaptados a la edad pediátrica, lo que ha permitido emplear con seguridad técnicas que desde hace un tiempo ya eran utilizadas en adultos con eficacia (ureteroscopia, litotricia extracorpórea con ondas de choque).
