



Advances in the diagnosis of rare genetic diseases and growing knowledge of the genes that cause them have allowed the exponential development of advanced therapies. Today, there is a therapeutic armamentarium that, while limited, was unthinkable years ago and is constantly evolving. Given the complexity of their mechanisms of action, the feasibility of clinical trials and the authorization by regulatory agencies, advanced therapies need to be investigated for the management and follow-up of pediatric patients. Ongoing pharmacovigilance of these therapies is also necessary to evaluate patient responses and outcomes. At this stage, novel phenotypes of disease emerge that had never been treated before. Prognostic factors and specific indications are also established, underscoring the importance of assessing each patient based on their particular characteristics, in adherence to the principles of the personalized medicine approach. The aim of this article is to provide a comprehensive summary of concepts and strategies as an introduction to the topic of advanced therapies for genetic diseases, taking into account their broad scope and their ongoing evolution and advancement.
Con los avances en el diagnóstico de las enfermedades raras de origen genético y el conocimiento cada vez mayor de sus genes responsables, las terapias avanzadas para estos trastornos se han ido desarrollando exponencialmente. Actualmente existe un arsenal terapéutico, si bien limitado, impensable hace años atrás y que está en constante evolución. Los niveles de complejidad en sus mecanismos de acción, la posibilidad de ensayos clínicos y aprobación por las agencias reguladoras hacen que las terapias avanzadas deban ser conocidas para el manejo y seguimiento pediátricos. También debe haber una constante farmacovigilancia a las que deben estar sometidas para ver la evolución y respuesta de los pacientes. A partir de estos puntos, aparecen entonces nuevos fenotipos en enfermedades que nunca habían sido tratadas con anterioridad. También se establecen factores pronósticos e indicaciones específicas, enfatizando la importancia de que cada paciente sea estudiado con sus características propias, de acuerdo con los patrones actuales que definen la medicina personalizada. Este trabajo pretende resumir, de manera representativa, conceptos y estrategias para introducir el tema de terapias avanzadas para las enfermedades genéticas considerando su amplitud y permanente evolución y progreso.
Medical genetics and therapies for genetic diseases have gained significant momentum in various spheres, from European directives to health care plans, health care provider groups, and patient associations. For a long time, the approach to the management of these diseases was primarily clinical. Today, thanks to advances in our understanding of their molecular underpinnings beyond basic research, there is progress in their accurate clinical and molecular diagnosis, comprehensive and multidisciplinary management, and treatment options. In this regard, the development of advanced therapies has revolutionized the current approach, serving as an example and a “mirror” for applying or promoting this field of research in other genetic diseases. Never before has so much attention been paid to genetic diseases that are disparate from an etiological and clinical point of view, but for which many treatment strategies may overlap.1
When a rare genetic disease is suspected or diagnosed based on the clinical manifestations, the next step is to confirm the diagnosis through genetic testing. Once it is confirmed, it may be possible to offer these patients specific treatment according to the affected gene and the molecular disorder through advanced therapies that act on DNA, RNA, or protein, depending on their effectiveness and availability. Despite these advances, adequate communication with the patient, as well as adequate information, remain essential to ensure that these therapies are carried out in accordance with good clinical practice and that expectations are managed appropriately.2,3
Preparation of clinical trials and therapeutic approaches to genetic diseasesGenetic diseases and treatmentAs is the case in other diseases, in order to investigate treatment options, it is necessary to understand the natural history of the disease, defined as its typical course, progression and outcomes in absence of any form of medical intervention or treatment. Any knowledge we have about the natural history of each disease for which accurate diagnosis is available will provide guidance in organizing interventions in the management of the different stages of the disease and its potential complications in order to improve the design of potential clinical trials.
The low frequency of these diseases must be taken into account, as it determines the stratification of cases for treatment or data analysis. Thus, many of the interventions and therapeutic options are based on studies involving few patients, and multicenter studies, both national and international, are generally necessary.
In some cases, medications that are used in the management of rare diseases may be known before their potential use or indication in these diseases is established. Thus, we have both orphan drugs and drugs used through expanded access, categories that are not mutually exclusive.
Orphan drugsOrphan drugs are treatments that are used in only a few people with a specific disease and, in general, support from the pharmaceutical industry for their development is more complex, leading to very high market prices compared to the development of treatments for other diseases. A significant proportion of approved advanced therapies are designated as orphan drugs, a status that also entails special considerations in obtaining authorization from regulatory agencies and health authorities.
Compassionate useCompassionate use or expanded access refers to the prescribing or administration of a drug to a patient before the drug is officially authorized for that particular indication. The Spanish Agency of Medicines (Agencia Española del Medicamento, www.aem.es)4 regulates expanded access to drugs that are still under development or awaiting authorization. Thus, there are three specific scenarios for compassionate use:
- 1)
Request to use an investigational drug outside of a clinical trial.
- 2)
The request for a drug that is not authorized in Spain (marketed in a different country).
- 3)
Prescribing of an authorized product for use in a way that is not described in the Summary of Product Characteristics.
Drugs used through expanded access are not used for investigational purposes and generally their indication is not subject to clinical trials, but rather is authorized in a particular instance when no other treatment is available.
Clinical trialsClinical trials are an important part of medical research that helps define treatments and protocols for different diseases.
When designing a clinical trial, two fundamental questions must be addressed: what will be tested, and which trial phase is appropriate. Clinical trials proceed through distinct phases:
- •
Early phase 1 trials, the preclinical exploratory phase in which the drug is investigated in the laboratory and in animal models of disease.
- •
Phase I trials focus on toxicity and the safe dosage range. They are usually conducted in healthy volunteers but may be conducted in people with the disease depending on the features of illness and the type of treatment being developed.
- •
Phase II trials focus on dose adjustment and pharmacokinetics and observation of potential therapeutic effects. These are generally pilot studies, that is, in a small number of patients and without placebo controls.
- •
Phase III trials involve a larger number of patients, using blinding whenever possible, and are conducted to assess efficacy on a large scale. These are considered pivotal trials, as they provide the most reliable data and can determine whether development continues and future marketing authorization is likely. Most of the data submitted to regulatory agencies are collected in this phase.
- •
Phase IV trials occur after the drug is authorized for marketing, and they provide definitive, long-term evidence on the drug’s effectiveness and adverse effects after the drug is marketed. These real-world data must be evaluated carefully, as they often inform decisions about whether to maintain or modify prescribing practices.5
With regard to what is tested in a clinical trial, it is not necessarily drugs or medications. Thus, it is also possible to test non-pharmacological interventions such as exercise, rehabilitation, psychotherapy, or dietary measures. When it comes to drugs, in addition to drug development, trials may be conducted to investigate drugs that are already marketed for a different use; new drugs to demonstrate their efficacy for a specific indication; improved drugs; and finally, a combination of drugs (eg, a new one with one that is already available and authorized).
In the case of genetic diseases, therapeutic strategies tested in clinical trials can be classified into four categories based on their level of intervention:
- 1)
Independent of the gene/pathophysiological mechanism (treats manifestations and complications)
- 2)
Acting on substrates or fundamental elements of the disease mechanism and its consequences (e.g., weak or atrophied muscle in motor neuron disease)
- 3)
Modifying genetic mechanisms (modifying or influencing the specific molecular basis, for example, promoting exon inclusion or inhibiting a toxic product)
- 4)
Replacing the involved gene or protein (depending on the product’s biodistribution)
Since most rare diseases are genetic in origin, it is essential to identify the involved gene or genes. These diseases may be caused by more than one gene, a phenomenon known as genetic heterogeneity. In general, the diagnostic process assumes that the disease at hand involves a single gene and is caused by a variant or variants of this gene. However, in some cases, other genes or modifying factors need to be considered as contributors to the specific phenotype or clinical manifestations.6 We also ought to remark on the importance of having measures or markers allowing to determine how the natural history of the disease, to the extent it is known, is modified by the delivered treatments or interventions. Thus, there are biological markers, commonly referred to as biomarkers, that offer objective, accurate and reproducible measures of health or disease status. In other words, a biomarker is a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes or responses to an exposure or intervention. They are used as a diagnostic and prognostic tool and to predict disease progression and outcomes. There are also predictive and pharmacodynamic markers that help assess the response to a specific drug.
Monitoring biomarkers may also be available for follow-up in genetic diseases. These biomarkers may be the products of the involved gene: RNA, the protein itself or its metabolites. They may also be other proteins or metabolites, present in blood (eg, creatinine) or cerebrospinal fluid (eg, neurofilaments) that are independent of the causative gene or genes but whose changes may be associated to the response to or efficacy of a drug or treatment under investigation. Last of all, it may also be necessary to monitor changes in drug substrates or the tissue considered most important in relation to treatment response. This may entail performance of histological analysis (eg, of muscle biopsy specimens) or imaging techniques, such as MRI or sonography.7
Definition of advanced therapiesOne of the possible definitions of advanced therapies is the one given by the Agencia Española del Medicamento,8 “medicinal products for human use based on genes (gene therapy), cells (cell therapy) or tissues (tissue engineering) and include products of autologous, allogeneic or xenogeneic origin”. It would also encompass combined advanced therapy medicinal products, which by definition necessarily include one or more advanced therapy products. These products enable new therapeutic strategies, and their development creates opportunities for diseases that currently lack effective treatments. The marketing authorization for these products involves submitting an application to the European Medicines Agency,9,10 which in 2009 instituted the Committee of Advanced Therapies (CAT) to draft opinions on these applications. The EMA broadly defines “gene therapy medicinal product” as a biological product that contains an active substance which contains or consists of a recombinant nucleic acid used in or administered to human beings with a view to regulating, repairing, replacing, adding or deleting a genetic sequence and whose therapeutic, prophylactic or diagnostic effect on the host genome relates directly to the recombinant nucleic acid sequence it contains, or to the product of genetic expression of this sequence. Of note, stemming from the circumstances of the COVID pandemic, the definition of gene therapy medicinal product excludes vaccines against infectious diseases, as, in principle, they do not modify the genome of the recipient.
In addition, the EMA defines “somatic cell therapy medicinal product” as a biological medicinal product that contains or consists of cells or tissues that have been subject to substantial manipulation so that biological characteristics, physiological functions or structural properties relevant for the intended clinical use have been altered, or of cells or tissues that are not intended to be used for the same essential function(s) in the recipient and the donor and that is presented as having properties for, or is used in or administered to human beings with a view to treating, preventing or diagnosing a disease through the pharmacological, immunological or metabolic action of its cells or tissues.
Lastly, the EMA defines “tissue-engineered product” as a product that contains or consists of engineered cells or tissues that may contain cells or tissues of human or animal origin and is presented as having properties for, or is used in or administered to human beings with a view to regenerating, repairing or replacing a human tissue. The cells or tissues may be viable or nonviable. It may also contain additional substances, such as cellular products, biomolecules, biomaterials, chemical substances, scaffolds or matrices.
These rather technical definitions can be expanded to take into account aspects of the disease itself, developing definitions and approaches that are more biological than technical, as explained below.
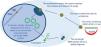
Biological context of gene therapies and gene therapy productsBeyond the technical definition of advanced therapies, the biological context of specific therapies for rare genetic diseases includes effects or modifications at the level of genes (Table 1), RNA, and protein, levels of action that are frequently complementary or lead to the application of these products as monotherapy or in combination therapies. Fig. 1 summarizes the areas of action of the most prominent therapies.
Types of gene therapy approaches.
| Possible gene therapy approaches | |||||
|---|---|---|---|---|---|
| Type of therapy | Definition | Target | Example | Therapeutic potential | Examples of diseases or experimental applications |
| Gene transfer | Deliver a functional copy of a mutated or missing gene to perform its normal function | Cell nucleus (eg, as episome) | Viral vector, usually AAV + healthy transgene (cDNA) | Replacement with a gene that restores the altered or lost function | Spinal muscular atrophy, Duchenne muscular dystrophy11,14 |
| Gene editing | Make a specific change in the sequence of the gene, without replacing it | Specific sequence of nuclear or mitochondrial DNA | Crisp-Cas9 | Any gene defect in the body | Sickle cell disease and beta thalassemia11,14 |
| Base editors | |||||
| Gene additiona | Introduce a new gene that is different from the altered gene in the body to act on a specific aspect of the mechanism of a usually multifactorial or complex disease | Cell nucleus, target organ or tissue | Viral vector + gene that produces antibodies, modifies signaling pathways, or produces neurotrophic factors | Supplement other therapeutic agents or difficult to treat diseases | Oncolysis or tumor cells (eg, melanoma), SERCA2a (calcium handling protein) in different heart diseases, transfer of antibodies in infectious diseases (eg, HIV)11 |
| Gene inhibition | Inactivate a mutated gene that results in a toxic product (eg, RNA blocking) | DNA or specific transcript | RNA interference | Blocking transcription or toxic RNA | Myotonic dystrophy, amyotrophic lateral sclerosis20 |
| Gene modification | Modifies gene expression or the gene product without altering the gene sequence | Pre-mRNA | Targeted antisense oligonucleotides | Include or remove exons to improve protein product | Spinal muscular atrophy, Duchenne muscular dystrophy19 |
Wang y Gao11 present gene addition as a functional modification approach independent of the defect that produces the disease. In this type of therapy, the causative gene itself is not treated (that approach would involve replacement, not addition).

Levels of intervention in advanced therapies targeting specific genes and their products. It is possible to act on DNA, either replacing the gene in the double-stranded genome, editing the variant (star) or activating or inhibiting its transcription through molecules that modify histones and gene expression. It is also possible to act on pre-mRNA, a single-stranded copy of the DNA sequence containing exons (blue) and introns (grey). Splicing of pre-mRNA can be modified to include or exclude exons through antisense oligonucleotides or splicing modulation compounds. The mRNA only contains the exons and can be inactivated partially or totally either with interfering RNA molecules or toxicity-blocking compounds. Proteins can be modified either at the translational level, with read-through compounds that preclude reading of stop codons in nonsense mutations, inserting an amino acid at that position to generate a longer protein, or with post-translational modification compounds (explained in greater detail in the text). Abbreviations: HDAC, histone deacetylase; RNA, messenger RNA.
The transfer of a normal copy of a gene to replace the function of a pathological variant is commonly known as gene therapy, the most widely used name to refer to this process. By extension, gene editing and the modulation of a gene through genetic mechanisms are also generally referred to as gene therapy. We ought to highlight that, conceptually, gene transfer refers to the incorporation of a normal version of the gene without acting on the mutated gene. Since DNA is very inefficient at reaching cells and passing into the nucleus, the most efficient way to transfer a gene is through a vector. There are chemical vectors, most commonly cationic lipids that interact with the negatively charged DNA molecule, thus forming a complex to deliver the genetic content to cells. They may carry large molecules of DNA and they exhibit a low immunogenicity. There is increasing investigation of these vectors to increase their efficiency and versatility and to achieve lasting expression in human recipients. There are also viral vectors, which are very efficient in delivering DNA to cells and achieving long-term expression, although they may be highly immunogenic and can cause inflammatory reactions in certain organs.
Usually, viral vectors containing genetic material have three key components: a promoter, a transgene (the genetic material the vector delivers) and a termination signal. It may also contain inverted terminal repeats (ITRs), which serve as the origin of replication for the synthesis of the nucleic acid copy. Viral vectors are natural viruses whose genome has been modified to achieve the desired function. The newly introduced gene can replicate autonomously and is controlled based on how the vector was constructed. The elimination of certain viral components from the vector alters its infectivity. In other words, the vector does not replicate the same way nor triggers the same immune response as the wild-type virus. There are five main types of viral vectors: retroviral and lentiviral vectors (which can carry transgenes of up to 8 kb), herpes simplex virus-based vectors (HSV, containing 40–150 kb of genetic material), adenoviral vectors (8–30 kb) and adeno-associated virus vectors (AAV, up to 5 kb of genetic material). The first two, retroviral and lentiviral vectors, integrate into the genome, which can lead to disruption of the host genetic material and cause harm to the patient. In contrast, HSV-1, adenoviral and adeno-associated virus vectors do not integrate into the host genome and persist in the cell nucleus as extrachromosomal episomes (Fig. 2).11

Illustration of the mechanism of action of gene therapy for the defective SMN1 gene using AAV9. The vector is taken up by the cell via an endosome, which then breaks down when it reaches the cell nucleus and releases its contents. Note that the vector contains the cDNA of the gene, that is, only the exons, without the introns.
At present, AAV vectors are the leading platform for gene transfer in the treatment of several human diseases. Many aspects have been improved in this approach, increasing its popularity and contributing substantially to the growth of the experimental and clinical field of gene therapy. These episomes replicate autonomously within the nucleus, although independently of the host genome. Administered intravenously, they are distributed to all cells throughout the body and generally can cross the blood-brain barrier (Fig. 2).
The vector enters the cell via endosome, the endosome breaks down in the cytoplasm, and the vector reaches the nucleus, where it delivers the transgene, which forms episomes. If the cell replicates, the episomes (usually 3–6 per cell) are distributed as they would in a cell division process. Thus, the effect is diluted over time until, after a few successive divisions, some of the cells will not receive the episome. In contrast, when delivered to cells that do not replicate, such as neurons, the episomes remain for a prolonged period, although the current data is insufficient to verify their long-term functionality. The viral vector dose varies, but is calculated based on patient weight with limits based on whether the patient is older or an adult. Thus, the dosage is calculated in vector genomes per kilogram of body weight for systemic intravenous administration, for instance, doses of up to 3 × 1014 viral genomes per kg of body weight have been administered for treatment of spinal muscular atrophy.
In respect of the host’s immune response to vector administration, there are important aspects to consider, such as the dose that can generate an initial response in the host and whether the host carries pre-existing neutralizing antibodies resulting from previous infections or, in the case of infants, transferred from the mother (passive immunity). All these factors can affect the effectiveness of the treatment. At present, gene therapy with AAV9-SMN1 (Zolgensma) is authorized by the Food Drug Administration (FDA) and the EMA in patients aged less than 2 years with spinal muscular atrophy, and trials are underway to investigate its intrathecal administration in older patients.12 Several clinical trials are also underway to investigate AAV vectors for management of other neuromuscular and metabolic disorders, among other diseases, which are registered and can be consulted in the ClinicalTrials.gov database.
Gene therapy carries risks, and these need to be taken into account in the selection of the optimal treatment option for the patient. Some of the factors to consider are the severity of disease, the availability of other therapies and the age of the patient. Deaths resulting from serious adverse events of gene therapy have been reported, chiefly in relation to the vector dose, hepatotoxicity and angiopathy. Thus, several trials may be discontinued if such complications occur. In any case, it is important to be well informed and not to create unrealistic expectations when deciding on this type of treatment for patients with serious genetic diseases.13
Gene editingGene editing is based on the ability to make very specific changes to the DNA sequence of a living organism and can be used to modify an altered sequence. It is performed using enzymes, particularly nucleases, designed to bind to a specific DNA sequence and capable of cleaving the DNA strands to allow recombination between the existing DNA and the replacement DNA, resulting in the insertion in the genome of the delivered DNA. The key to this gene editing technology is a molecular tool known as CRISPR-Cas (clustered regularly interspaced short palindromic repeats and caspase 9).14
Unlike gene transfer, which entails delivery of a complete gene with normal function, in gene editing it is the host’s altered DNA that is modified to restore wild-type expression or remove the aberrant expression.
Precision gene editing is being investigated in clinical trials for cancer immunotherapy and treatment of viral infections and hereditary hematologic, metabolic and eye disorders. At present, these are very specific clinical applications that achieved a localized effect on somatic cells and not a generalized distribution throughout the body. Thus, it can be used in skeletal muscle, whose cells are postmitotic and multinucleated, which would allow repair of a subpopulation of nuclei potentially beneficial to the entire muscle fiber. It can also target the retina and the liver. Some of the main advantages of gene editing are its versatility, as it can target any specific sequence or cell type, that it can be used to edit single point polymorphisms, deletions, or duplications, and that it could potentially be applied to nearly all genetic diseases. Its main drawbacks, are that its efficiency may be low, that it may modify germ cells so that the edited sequence is passed on to future generations, that it may result in unintended off-target editing of genomic regions other than the desired one, or, in the case of on-target editing, generate genetic mosaicism, and, above all, that its long-term effects are currently unknown. However, there are more than 80 active clinical trials using CRISPR technology, and almost all applications in current clinical trials correspond to localized diseases or target specific organs. CASGEVY, the first gene editing therapy for sickle cell disease and beta thalassemia, has recently been approved at a cost of approximately €2 million per patient. It is an ex vivo therapy that involves editing of blood cells extracted from the patient that are later reinjected. In mitochondrial diseases with heteroplasmy, in which both the variant and the wild-type sequences are present in the same cell, the disease may manifest depending on the number of abnormal copies. In this case, mitochondrial DNA can be modified by delivering nucleases that specifically cleave and eliminate mutant mitochondrial DNA, an effect that has been observed both in heteroplasmic cells and animal models.
Base editors that make base changes via precise C>T or A>G transitions through chemical modification of the bases are currently being used in gene editing experiments and trials. There is also a third generation of caspases that, controlled through an RNA guide, can cleave a single strand (instead of two strands, like the first version of CRISP-Cas9).16 All of these innovations aim at reducing the risks and disadvantages and increase the efficacy of gene editing.
As in the case of gene therapy for spinal muscular atrophy, which involves systemic administration via intravenous injection, the extremely high price tag of all these therapies raises important questions about the feasibility of making them available to the entire patient population and questionable issues in relation to pricing schemes and the financial sustainability of health care systems.15Table 2 presents some of the high-cost therapies currently authorized for use in humans.
Gene therapies that use AAV approved for use in humans.
| Vectora | Capsidb | Disease | Year of approval |
|---|---|---|---|
| Voretigene neparvovec (Luxturna) | AAV2 | Retinal dystrophy | 2017 |
| Onasemnogene abeparvovec (Zolgensma) | AAV9 | Spinal muscular atrophy | 2019 |
| Etranacogene dezaparvovec (Hemgenix) | AAV5 | Hemophilia B | 2022 |
| Valoctocogene roxaparvovec (Roctavian) | AAV5 | Hemophilia A | 2023 |
| Delanistrogene moxeparvovec (Elevidys) | rh74 | Duchenne muscular dystrophy | 2024 |
| Eladocagene exuparvovec (Kebilidi/Upstaza) | AAV2 | Aromatic l-amino acid decarboxylase deficiency | 2024 |
Abbreviation: AAV, adeno-associated virus.
The “parvo” part of the name indicates that the vector is a parvovirus, a family of viruses that includes adeno-associated viruses. The suffix “vec” means that the component is a vector, a non-replicating virus used to transport genetic material.
The viral capsid protects the viral genome from degradation and facilitates entry into the host cell. It is formed by three viral proteins (VP1, 2, 3), whose different serotypes characterize the infectivity (which tissues it penetrates and whether it crosses the blood-brain barrier) and immunogenicity.
Epigenetics refers to anything that changes the expression of a gene without changing its sequence. For example, DNA methylation (addition of methyl groups to the DNA) and histone deacetylation (histones are proteins that interact with chromatin) impede access to the replication machinery so it cannot have access to the template to express the gene. These mechanisms can be modified through pharmacological agents, such as histone deacetylase (HDAC) inhibitors, widely used in cancer research and trials, although their applications in other diseases is still limited. One of the best-known HDAC inhibitors is valproic acid, an anticonvulsant that can also act as an epigenetic modifier on hundreds of genes and that has been used in several clinical trials in the field of spinal muscular atrophy due to its in vitro and in vivo capacity to increase expression of the SMN gene and the production of full-length SMN RNA transcripts in patients with this disease. The results of these trials were disappointing.17 However, it may be a useful addition to the therapeutic armamentarium as a coadjuvant for other disease-modifying therapies.
RNAThe function of genes is to carry information so that the body’s proteins can be synthesized through messenger RNA (mRNA). This protein synthesis process takes place in two stages: transcription, which consists of the synthesis of pre-mRNA from DNA, splicing of pre-mRNA and formation of mature mRNA; and translation, which consists of the synthesis of proteins by ribosomes using mRNA as a template.
Pre-mRNA and antisense therapyPre-mRNA includes exons and introns and undergoes splicing to remove the introns and join the exons together to form mature mRNA. It is synthesized using the antisense strand of DNA as a template. As a reminder, DNA is double-stranded with both strands running antiparallel: each strand has a 5′ end and a 3′ end, with the sense and antisense strands oriented in opposite directions. The single strand of pre-mRNA uses the antisense template to generate a 5′ to 3′ single-stranded sequence with the same directionality as the sense strand. The mature mRNA exits the nucleus and enters the cytoplasm, where ribosomes synthesize the corresponding polypeptide.
Antisense oligonucleotides hybridize with complementary sequences in single-stranded pre-mRNA. These oligonucleotides are short single-stranded molecules of 8–50 bases designed according to the target sequence. Upon binding to pre-mRNA, they modulate splicing by forming a double-stranded segment at the target site.
Although they do not cross the blood-brain barrier, antisense oligonucleotides achieve widespread distribution in the central nervous system (CNS) after intrathecal injection, making them very valuable as therapeutic tools in neuronal diseases.18
It is important to note that if the sequence targets the exon, the exon will be excluded from the mature RNA (exon skipping), as occurs with the dystrophin gene in Duchenne muscular dystrophy therapy. In contrast, hybridization with intronic regions that inhibit inclusion helps with exon inclusion. This has given rise to terms such as exonic splicing enhancer (ESE) or exonic splicing silencer (ESS) as well as intronic splicing enhancer (ISE) or intronic splicing silencer (ISS). This is the mechanism of action of the nusinersen oligonucleotide (Spinraza), of 18 bases, which specifically binds intronic splicing silencer N1 (ISS-N1) in intron 7 of the SMN2 gene. This inhibits binding of two splicing repressors in exon 7 (hnRNPA1 and A2), enabling the inclusion of this exon in the mature RNA. The drug was approved by the FDA in 2016 and by the EMA a year later for treatment of all forms of spinal muscular atrophy.19
RNA toxicityAnother treatment approach is to reverse or block the toxic effect of a specific RNA that is harmful to the cell. For example, this is being investigated in myotonic dystrophy caused by CTG triplet expansion (CUG in RNA) in the DMPK gene. Following transcription, the expanded CUG repeat in the DMPK mRNA sequester the MBNL1 splicing factor resulting in loss of function. The CUG repeat expansion also leads to a relative gain of function in another splicing factor, CUGBP1. The imbalance between MBNL1 and CUGBP1 results in aberrant splicing events that cause the characteristic symptoms of the disease. The expansions can be blocked with chemical agents, such as pentamidine, which bind the CUG repeats in competition with the splicing factors, thus neutralizing the effect of the repeats and restoring normal splicing.20
It is also possible to block or degrade RNA by means of RNA interference (RNAi). In this case, the intervention works on mature RNA, harnessing an existing biological process by which RNA molecules suppress gene expression of double-stranded RNA genes to regulate mRNA expression through complementary base pairing. An enzyme (Dicer) is used to cleave long double-stranded RNA molecules into several small interfering RNA (siRNA) molecules. One of the siRNA strands is loaded into a protein complex known as RISC (RNA-induced silencing complex) that identifies and cuts the aberrant mRNA, which is then degraded by the cell’s RNA decay machinery. This blocks expression of the aberrant gene. When it comes to its application for human use, a drug based on AAV-RNAi for treatment of Huntington disease is currently undergoing Phase I/II trials (NCT04120493). It uses an AAV5 to deliver a microRNA that targets the huntingtin protein mRNA. These studies will probably yield important safety and feasibility data that may also be relevant for the use of this approach in the treatment of other diseases involving triplet expansion or RNA toxicity
ProteinsReplacement/delivery of the affected proteinReplacement therapies, whether they replace the gene or its products, may seem like the simplest and most obvious solutions at first glance. However, the replacement mechanisms are not straightforward, as many factors must be taken into account, including molecular weight, route of administration, tissue penetration, half-life, and the metabolism of externally administered proteins. Proteins are usually synthesized artificially (recombinant proteins) given the dangers of pathogen transmission when they are obtained from other sources. Some proteins are difficult to deliver to the target organ or cell, and systemic administration of proteins may also pose problems if they have a large molecular weight.
Translation-targeted strategiesThere are three key steps in the translation of RNA to protein by ribosomes: initiation, elongation and termination. Many neuromuscular diseases involve nonsense mutations in which an amino acid codon is replaced by a stop codon. These premature termination codons prevent the synthesis of the full-length product, resulting in a truncated protein that undergoes rapid degradation. In many cases, the mRNA carrying the stop codon is even degraded before being translated. It is possible to inhibit the reading of premature termination codons with compounds such as aminoglycosides, PTC124, and, more recently, Elox. These drugs have been tested to modify the effect of nonsense mutations through the addition of another amino acid at the site of the termination codon. The transfer RNA carrying the amino acid may not introduce the specific amino acid present in the normal protein, and in many instances transforms a truncated protein (resulting from the nonsense mutation) into a full-length protein that has an amino acid change at that point in the sequence, as if it were a, but this protein will have an amino acid change (as if the defect were a missense mutation). Thus, it has been observed that the use of PTC124 tends to result in the insertion of specific amino acids to replace the stop codon: Gln, Lys or Tyr if the stop codon is UAA or UAG, and Trp, Arg or Cys if the stop codon is UGA.21
Post-translational modificationOnce the protein is synthesized (if the disease mechanism allows the synthesis of an aberrant protein different from the normal one), post-translational modification and function may include transport of the protein to the membrane, forming a complex with other proteins, acting as a catalyst for a reaction, or functioning within a specific metabolic pathway. To perform some of these functions and increase or reduce its stability, the protein may have folds and signals, and its potential functions can be expanded by attaching additional functional groups, such as acetates, sugars (glycosylation) for tagging, recognition or phosphorylation, and disulfide bridges to change its shape. Finally, the protein is metabolized and degraded by different mechanisms through the lysosome, ubiquitins, or proteasome. In this regard, there are medications that target the chemical modifications or degradation of proteins of interest, making this a very promising field of study.22 As mentioned above, ClinicalTrials.gov is a database of publicly and privately funded clinical trials conducted worldwide, where it is possible to consult all medications of this type that are being investigated in humans.
Levels of prevention in genetic diseasesWith the advent of advanced therapies for genetic diseases, the term disease-modifying therapies (DMTs) has been introduced to refer to those that radically change the course and prognosis of the disease. The first approach to the prevention of disease involves treating all patients who manifest the disease (tertiary prevention). Its implementation primarily affects management and follow-up care, and also gives rise to alternative phenotypes or courses of disease in response to the delivered treatment.23 Although treating the disease once symptoms appear is an improvement, preventive measures are also implemented with the aim of treating the disease before symptoms develop (secondary prevention). One of the current universal secondary prevention measures is newborn screening for diseases for which treatment is available that can prevent or minimize their manifestations, improving quality of life and reducing the social and health care burden of the disease. Although screening has traditionally involved biochemical methods (eg, screening for phenylketonuria or hypothyroidism), genetic screening programs (for severe immunodeficiency, spinal muscular atrophy) are gradually being introduced. Finally, regardless of the screening method, most of the diseases tested for have a genetic cause. That is why some countries (Israel, Netherlands) are conducting population carrier screening for some genetic disorders, based on their incidence and carrier frequency, to provide genetic counseling and offer the appropriate reproductive options to each couple. This leads to influencing the incidence and prevalence of the disease. Health care plans and systems have to weigh the advantages and disadvantages of each intervention to integrate them in the service portfolio in a way that fits the specific population. Clinical research in advanced therapies for genetic diseases is the positive result of a constant commitment to basic translational and clinical research. Such proof-of-concept trials are greatly helping the development of therapies for spinal muscular atrophy, Duchenne muscular dystrophy, cystic fibrosis, and other complex and heterogeneous genetic diseases. In the field of advanced therapies and genetic diseases, there will certainly be no single optimal solution or approach, given the pathophysiology and progression of each disease. It will also be necessary to determine when treatment can be started, which may be as early as the prenatal period.24,25 The special characteristics of each patient should also be considered, beyond the therapies that are available, to clearly establish which types of patients are being prescribed a specific therapy. It is important to consider that combining different complementary treatment mechanisms or strategies can result in an effective therapeutic approach.
ConclusionsRare genetic diseases contribute significantly to morbidity, mortality, and overall health care costs. They are serious disorders that require early diagnosis and proactive measures for monitoring and treatment. In this regard, genetic techniques play a crucial role as diagnostic tools and in the development of advanced therapies that, as they become safer and more effective, become increasingly feasible in clinical practice, although their costs may affect access to them as well as equity. The development of advanced therapies for a given disease requires knowledge of its natural history and genetic underpinnings to validate their safety and efficacy in controlled clinical trials. The next step in the application of advanced therapies is to pursue initiation of treatment at the earliest possible stage. In this regard, treatments for some genetic diseases known to start manifesting in utero may be available in the near future.24,25 Today, replacing a gene with gene therapy and modifying RNA with antisense therapy is already a reality. We know that these advances, applied to patient care, are effective, and that, while they pose new challenges in terms of monitoring and assessing patient response and outcomes, they motivate health care providers and improve the quality of life of patients and their families.
The author has received fees as a consultant for Novartis Gene Therapies, Biogen, Biologix, Cytokinetics, Novartis, and Roche, and has received research funding from Biogen/Ionis and Roche.
