


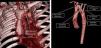
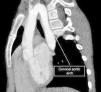

We present the case of a girl aged 9 years with DiGeorge syndrome referred to our hospital due to suspicion of right aortic arch. In the physical examination, the patient exhibited mild dyspnea, cough and occasional choking, with a palpable cervical pulse. The computed tomography (CT) scan of the heart revealed a complex vascular anomaly: a right-sided aortic arch extending cranially to the level of the right thoracic inlet, forming a cervical aortic arch (CAA) (Figs. 1 and 2, Appendix B video 1). The first branch of the aorta was the left common carotid artery, followed by the right subclavian artery, the left common carotid and the left subclavian artery (LSA). The LSA arose from a Kommerell diverticulum (Fig. 3, Appendix B video 2), which, combined with the ligamentum arteriosum, formed a complete vascular right that compressed the trachea (Fig. 3). Cervical aortic arch is an infrequent anomaly in the development of the aorta, occurring in fewer than 1 in 10 000 live births, characterized by an elongated aortic arch extending at or above the medial ends of the clavicles.1 This condition is associated with aneurysms (occurring in up to 20% of cases), coarctation of the aorta, congenital heart defects, Turner syndrome and DiGeorge syndrome.1,2 Given the presence of a complete vascular ring, surgery was the chosen treatment.3
Impact factor
The Impact Factor measures the average number of citations received in a particular year by papers published in the journal during the two preceding years.
© Clarivate Analytics, Journal Citation Reports 2025
Impact factor 2024
2.1
View more metrics



