



The congenital abnormalities of kidney and urinary tract (CAKUT) are disorders with a high prevalence in the general population, with urinary tract dilations being the most frequent. CAKUT also account for the most important cause of chronic kidney disease in childhood.
This paper focuses on the role of the primary care paediatrician in the diagnosis, assessment, and follow-up of children with CAKUT, with special emphasis on the associated urinary tract infections, the progression towards chronic renal failure, and the genetic basis.
Las anomalías nefrourológicas congénitas (CAKUT) son alteraciones con una alta prevalencia en la población general; de ellas, las más frecuentes son las dilataciones de la vía urinaria. Suponen además la causa más importante de enfermedad renal crónica en la edad infantil.
En este artículo se hace especial énfasis en el papel del pediatra de Atención Primaria en la valoración y el seguimiento de los niños con CAKUT, fundamentalmente en lo que hace referencia a las infecciones urinarias asociadas, a la progresión hacia la insuficiencia renal y a su base genética.
The purpose of this article is not to review the diagnostic and treatment methods that need to be used in the different forms of congenital anomalies of the kidney and urinary tract (CAKUT), as these procedures are usually performed in hospital paediatric nephrology and urology units, but to discuss current aspects of this pathology, as it is relevant to paediatricians. As a whole, the CAKUT constitute a group of disorders of great relevance in clinical practise due to their high prevalence, being a cause of chronic renal disease, and to important breakthroughs and advances in the knowledge of its pathogenesis and clinical expression that have occurred in recent years.
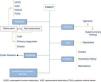
The term CAKUT includes a considerable number of diseases caused by defects in the morphogenesis of the urinary system, including alterations in the number, size and/or position of the kidneys, obstructive or non-obstructive dilatation of the urinary tract, and dysplastic kidney lesions, including cystic disorders. They may occur in isolation or in the context of a syndrome. Fig. 1 offers a graphic representation of the general classification of the various types of CAKUT (we ought to note that the duplex renal collecting system must be considered a variant of normality unless it is associated with other malformations). Some authors include autosomal dominant or recessive forms of polycystic kidney disease as well as nephronophthisis in this umbrella. Although strictly speaking these diseases can be considered CAKUT, they require a distinct diagnostic and therapeutic approach and they do not present many of the clinical and outcome features that are generally shared by the CAKUT, which is why they will not be discussed further in this article.
In our region, CAKUT are generally detected before birth, and they account for approximately one third of prenatal ultrasound abnormalities. Some type of CAKUT is found in 5–10 of 1000 live newborns, and they are the most commonly found type of malformation detected in humans.1 The most frequent among these malformations involve the dilatation of the urinary tract. We ought to note that while dilatations of the urinary tract are easy to detect in the prenatal period, it is common for small kidneys (renal hypoplasia) to go unnoticed in ultrasound examinations during pregnancy. This should be taken into account when establishing the clinical indications for renal ultrasound in postnatal life.
This article aims to be useful to paediatricians in answering the following questions about CAKUT: Do they carry a higher risk of urinary tract infection (UTI), and what are the characteristics of these infections? What are the long-term renal outcomes? What do we know of their genetic basis and how should families be counselled in regards to inheritance and the risk of recurrence in future children?
Urinary tract infectionIt is well known that UTIs are more frequent in female individuals, and thus 7% to 8% of girls and 2% of boys have at least one UTI in the first eight years of life.2 The incidence of febrile UTI is highest in the first year of life in both sexes,2 and the higher frequency in males during the neonatal period and the first months of life is related to the higher frequency of CAKUT in boys than in girls.3 Infants with dilatation of the urinary tract are at higher risk of developing acute pyelonephritis in the first year of life, so that the probability of being hospitalised for this reason is approximately twelve times that of children without hydronephrosis.
While the use of continuous antibiotic prophylaxis for the prevention of UTIs is still being debated, a review of 21 studies published between 1990 and 2010 on the incidence of UTIs in children aged less than 2 years with antenatal hydronephrosis concluded that antibiotic prophylaxis does not reduce the rate of UTIs in patients with low-grade hydronephrosis (ultrasound findings showing absent or minimal dilatation of the calyces, absence of thinning of renal parenchyma, and/or postnatal anteroposterior renal pelvis diameter between 4.0 and 14.9mm), approximately 2.5% of whom received a UTI diagnosis regardless of whether or not they had received prophylaxis, while significantly reducing the risk of UTI from 29% to 15% in patients with grade III and IV hydronephrosis and/or an anteroposterior renal pelvis diameter equal to or greater than 15mm, although the level of evidence was low.4 Thus, infants with high-grade antenatal hydronephrosis may benefit from antibiotic prophylaxis to reduce the probability of contracting a UTI.
We must also highlight that Escherichia coli (E. coli) is less prominent as a causative agent of UTIs in paediatric patients with CAKUT than in the general population,5 which must be taken into account when it comes to the initiation of therapeutic or prophylactic measures. Another important consideration is that while the probability of a UTI or even recurrent UTIs causing chronic renal disease is very low,6 the association of acute pyelonephritis and obstructive uropathy carries a high risk of permanent renal damage unless antibiotic treatment is initiated promptly and the obstruction is eliminated, allowing the drainage of infected urine from the involved kidney.
Renal function outcomesThe CAKUT are the cause that accounts for the highest percentage of chronic renal disease in all paediatric case series, and if we include cystic diseases in the term, they cause nearly three fourths of all cases of chronic renal failure in children. Fifty-six percent of the cases of chronic renal failure stages 2–4 (not undergoing dialysis) documented in the registry of the Asociación Española de Nefrología Pediátrica (Spanish Association of Paediatric Nephrology) are due to structural renal or urologic malformations, and this percentage is similar to those found in European and North American case series, in which the percentage of chronic renal disease due to CAKUT ranges between 48% and 59%.7 The risk of chronic renal disease depends on the number of functioning nephrons at birth, and thus on the history of preterm birth, the degree of renal dysplasia and the presence of bilateral or unilateral involvement, as well as acquired nephron loss, which in children is mostly due to the co-occurrence of upper UTI and/or persistent obstruction (Fig. 2).
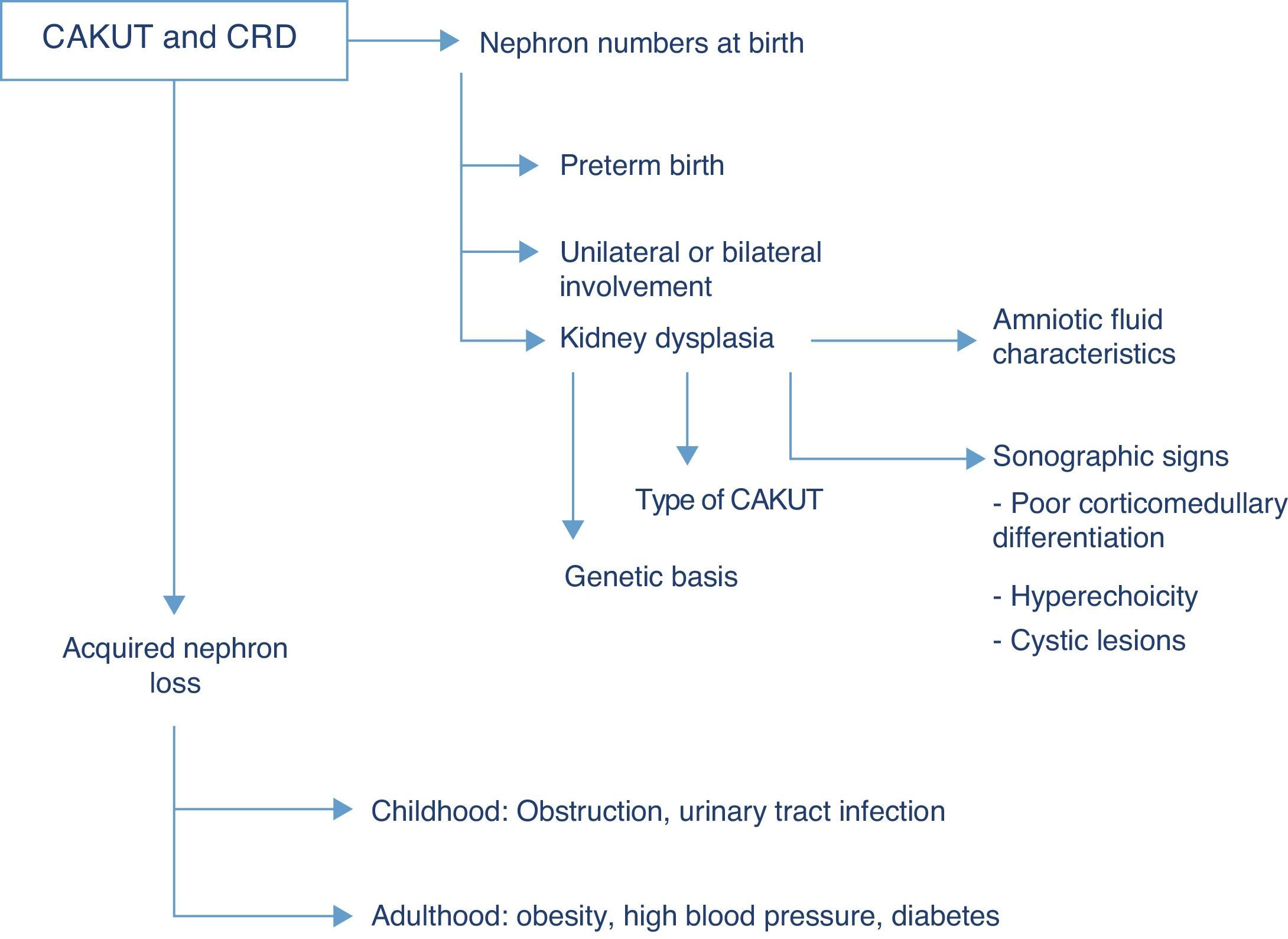
While the term dysplasia refers to anatomic and histopathological characteristics of the tissue at hand, different clinical findings may suggest its potential presence, such as hyperechoic regions in the renal parenchyma, diminished corticomedullary differentiation or the detection of renal cysts in ultrasound imaging. Before birth, at 20 weeks of gestation, foetal urine already amounts to more than 90% of the total volume of amniotic fluid, so the finding of oligohydramnios past this gestational age is an excellent indicator of abnormal renal function in the foetus.8 Generally speaking, sodium and chlorine concentrations higher than 90mEq/L and osmolalities below 210mOsm/kg H2O in amniotic fluid suggest renal tubule involvement and a poor prognosis, although none of these biochemical parameters predicts postnatal renal function with sufficient clinical certainty.9 In addition to the congenital renal damage caused by dysplasia, the nephropathy associated to CAKUT usually originates in the renal interstitium, so when CAKUT end up causing chronic renal failure, the latter usually manifests with preserved diuresis or even polyuria. The development of microalbuminuria is considered a marker of glomerular damage, already suggested by the detection of sustained proteinuria unless it involves low-molecular-weight proteins.
The presence of kidney failure from birth or the first weeks of life is a predictor of poor prognosis in regards to renal function, although in some of these children glomerular filtration, corrected for 1.73m2 of body surface area, may improve with the rapid growth of the body during the first year of life. Another important consideration is that the impairment of renal function may worsen during puberty, as the affected kidney or kidneys do not grow in proportion to the rest of the body. Similarly, a partial obstruction of the urinary tract may be exacerbated by the pubertal growth spurt and cause an obstructive uropathy, compromising the function of the dilated kidney. These circumstances and possible progressions of disease must be kept in mind when planning the clinical follow-up of these patients as they enter the pubertal period.
Genetic basisThe pathogenesis of the different diseases under the CAKUT umbrella is complex, consistent with the complex mechanisms involved in the embryonic morphogenesis and development of the urinary system. The CAKUT occur more frequently in males than in females, and there are cases of family aggregation. They may be part of multiorgan processes in single-gene disorders with dominant or recessive inheritance, such as branchiootorenal syndrome, Kallmann syndrome, Fraser syndrome, Ehlers–Danlos syndrome and Townes-Brocks syndrome, among others.
There are also monogenic forms of CAKUT in which the renal phenotype is the sole or predominant feature. Among the various genes that may be involved, the most frequent ones are HNF1β and PAX2. Mutations in the PAX2 gene cause renal hypoplasia associated with coloboma and deafness.10 Also, mutations in this gene have been associated with isolated renal hypoplasia and dysplasia and with multicystic dysplastic kidney.11 In recent years, defects in the HNF1β gene that encodes the homonymous transcription factor (hepatocyte nuclear factor 1β) have been shown to cause various developmental abnormalities, and some authors have even proposed the term HNF1β-associated disease.12 This transcription factor is involved in the organogenesis of the kidneys, urinary tract, liver and pancreas, and there is evidence that mutations in this gene account for up to 10% of CAKUT cases.13 The presence of underlying CAKUT-causing HNF1β mutations should be suspected in cases of bilateral kidney lesions with detection of renal cysts of unknown origin associated with a positive family history of diabetes, pancreatic hypoplasia and electrolyte abnormalities such as hypomagnesaemia and hyperuricaemia.
Thus, in addition to investigations directly related to renal and urologic structural abnormalities, the evaluation of a patient with CAKUT should include the purposeful exploration of extrarenal manifestations and a detailed family history to help identify the underlying molecular cause and facilitate the genetic counselling of affected families.
Key pointsThe following key points must be taken into consideration by the paediatrician in the diagnosis and follow-up of a child with CAKUT:
- –
The term CAKUT encompasses a heterogeneous group of disorders that are very frequent, are associated with UTI and constitute the major cause of chronic kidney disease in the paediatric age group.
- –
If a CAKUT has not been detected prenatally, it should be suspected in male infants with UTI, especially those with pyelonephritis that require hospitalisation and with infections caused by pathogens other than E. coli. This must be taken into account not only to establish antibiotic treatment but also during follow-up, as upper urinary tract infections carry a high risk of permanent renal damage in the context of obstructive urinary disease, and thus their course must be monitored closely.
- –
The correct diagnosis of CAKUT requires a detailed history taking, including family history of kidney problems and diabetes, and the search for associated extrarenal malformations: coloboma, deafness, hypomagnesaemia, hyperuricaemia, etc.
- –
The follow-up of these patients must avoid factors that may aggravate the existing congenital renal damage, with an emphasis on the prevention and treatment of acute pyelonephritis, the elimination of urinary tract obstruction, and the promotion of healthy lifestyle habits. In this regard, it is particularly important to consider diseases that involve sustained hyperfiltration by residual functioning nephrons, as this heightens the risk of glomerulosclerosis. The development of pathological microalbuminuria and more so of sustained proteinuria are markers of glomerular damage.
- –
Antibiotic prophylaxis in children with hydronephrosis has only been demonstrated to reduce the risk of UTI in patients with high-grade disease.
- –
During the pubertal growth spurt, the impairment in renal function may be exacerbated, so the primary care physician, who will be in charge of providing continuing care, should be informed in detail of the clinical condition of the patient to ensure an appropriate follow-up during this stage of life, especially in cases of renal or urologic disorders such as renal hypoplasia or urinary tract obstruction.
The authors have no conflict of interests to declare.
Please cite this article as: Palacios Loro ML, Segura Ramírez DK, Ordoñez Álvarez FA, Santos Rodríguez F. Anomalías nefrourológicas congénitas. Una visión para el pediatra. An Pediatr (Barc). 2015;83:442.e1–442.e5.
Previous presentation: the results of this study were presented in part at the Reunión de la Sociedad de Pediatría de Asturias, Cantabria y Castilla y León, April 10–11, 2015.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals