










Diversos estudios estiman que el retraso mental afecta a un 1-3% de la población, y en cerca de un 50 % de los casos se desconoce la etiología. La incertidumbre sobre la etiología, y la recurrencia, hacen que la prevención del retraso mental presente graves repercusiones de tipo terapéutico, social e incluso económicas.
La clave principal es lograr un diagnóstico preciso, probando una hipótesis clínica mediante la realización de las pruebas genéticas adecuadas. Debido al creciente desarrollo de la tecnología en el campo de la genética, y a la disponibilidad de nuevas pruebas, en este artículo se revisan e integran los criterios establecidos en las guías consensuadas de diferentes sociedades científicas (pediátricas, neurológicas y genéticas) respecto a su utilización en el diagnóstico del retraso mental y del retraso del desarrollo.
Different studies show that mental retardation affects 1-3 % of the population, and in about 50 % of the cases the aetiology is unknown. The uncertainty on the aetiology, and recurrence, means that prevention of mental retardation can have serious, therapeutic, social, and even economic repercussions.
The key is to obtain an accurate diagnosis, proving a clinical hypothesis by the accomplishment of the most suitable genetic tests. Due to the increasing development of the technology in the field of the genetics, and the availability of new tests, this article reviews the criteria established in the practice guidelines from different scientific societies (paediatric, neurological and genetic) with respect to their use in diagnosis and integrates them from the point of view of their use in mental retardation and developmental delay.
La expresión “retraso del desarrollo” implica un déficit de aprendizaje y adaptativo que puede ser significativo y predecir una discapacidad posterior cognitiva o intelectual. La prevalencia del retraso del desarrollo se estima comprendida entre el 1 y el 3 %. El tipo de retraso del desarrollo es un paso preliminar importante, dado que influye sobre la vía de investigación posterior. Su clasificación respecto a la afectación es clínicamente subjetiva, puesto que se reserva para niños habitualmente menores de 5 años en quienes no puede realizarse la evaluación del cociente intelectual (CI).
A edades tempranas, los retrasos del desarrollo catalogados como leves o moderados pueden ser transitorios y carecer de capacidad de predicción respecto al desarrollo de un posterior retraso mental u otras discapacidades cognitivas, en especial el retraso del desarrollo de un solo dominio (retrasos motores o retrasos del lenguaje). Cuando estos signos se presentan aislados de una forma grave, y en particular si se presentan asociados con otras discapacidades, dismorfias y/o malformaciones congénitas, puede ser importante llevar a cabo una revaluación clínica para decidir el mejor abordaje en la evaluación diagnóstica. En la práctica, debido a la falta de recursos en la estructura sanitaria actual, muchos niños continúan con un diagnóstico de retraso del desarrollo desde los 5 años en adelante.
La expresión “retraso mental” se aplica a niños mayores de 5 años cuando han sido examinados mediante un cuestionario de evaluación del CI, y éste es inferior a 70. Se caracteriza por una funcionalidad cognitiva significativamente limitada, acompañada de limitaciones adaptativas en dos o más de las siguientes áreas: habilidades sociales, sociabilidad, comunicación, autonomía, salud, trabajo y aprendizaje. El retraso mental se divide generalmente en: leve (CI, 50–70), moderado (CI, 35–50) y grave (CI, 20–35). Los casos con un CI inferior a 20 son catalogados como con retraso profundo1–3.
La etiología del retraso mental/retraso del desarrollo es compleja, e incluye factores ambientales y genéticos, y a la hora de establecer una etiología final estos factores pueden presentarse solos o en combinación con otros. Las principales causas pueden resumirse en las siguientes: anomalías cromosómicas, anomalías del desarrollo del sistema nervioso central (SNC), teratógenos ambientales, retraso mental sociocultural, complicaciones de la prematuridad, enfermedades monogénicas conocidas, causas sindrómicas y enfermedades metabólicas o endocrinas. Se estima que las causas genéticas podrían llegar a estar implicadas en un 50 % de los casos de retraso mental o del desarrollo, aunque es, hoy día, poco practicable llegar a estos niveles de confirmación diagnóstica.
EVALUACIÓN CLÍNICA EN PACIENTES CON RETRASO MENTAL O DEL DESARROLLOCobra gran importancia el proceso de evaluación clínica en tanto que puede cambiar la dirección de los estudios posteriores a realizar. Para que el proceso sea eficaz, éste debe seguir un protocolo ordenado, que conste como mínimo de los pasos contenidos en la tabla 1.
Protocolo de evaluación clínica del paciente con retraso mental o del desarrollo
Historia clínica detalladaÁrbol genealógico detallado de tres generaciones y detalles sobre el desarrollo de todos los posibles individuos afectados: la anotación en la historia clínica debe estar acompañada por la interpretación de la herencia, si los antecedentes en la familia son suficientes o lo permiten
|
| Diagnóstico diferencial en el retraso mental o del desarrolloLos datos de la historia clínica, junto con la exploración física del paciente y la investigación de los datos extraídos utilizando la bibliografía existente, u otras herramientas, deben conducir a un diagnóstico diferencial, estableciendo una hipótesis diagnóstica |
| Elaboración de una hipótesis diagnósticaEn el proceso de llegar a elaborar una hipótesis diagnóstica clínica deben evaluarse todos los detalles con una metodología rigurosa, de ahí que diferentes consensos publiquen tablas y criterios de evaluación clínica en pacientes con retraso mental o del desarrollo18. Esta hipótesis deberá confirmarse diseñando un panel de pruebas diagnósticas complementarias que deben aplicarse |
| Pruebas complementarias y de confirmación diagnósticaLas pruebas diagnósticas de las que se dispone actualmente son numerosas y variadas (electroencefalograma, exploraciones de neuroimagen, cariotipo, entre otras); por esta razón, dependiendo de la hipótesis diagnóstica establecida, se diseñará un plan secuencial para su aplicación. Existen en la bibliografía numerosos estudios que permiten estandarizar cómo, y con qué criterios, deben ser utilizadas las peticiones de pruebas complementarias para abordar un diagnóstico diferencial concreto |
| Confirmación de la hipótesis diagnósticaCuando ésta se confirma, se obtiene un diagnóstico definitivo. Si esto no ocurre, es necesaria una revaluación clínica y establecer una nueva hipótesis diagnóstica |
Las pruebas genéticas deben indicarse cuando:
- 1.
Se haya realizado una exploración básica.
- 2.
Exista una historia familiar positiva de retraso mental o del desarrollo, de abortos o de mortinatos sin causa explicada.
- 3.
El retraso del desarrollo o mental se asocie con rasgos dismórficos, fenotipo conductual indicativo, regresión o consanguinidad.
- 4.
Se hayan descartado por completo otras causas.
Incluso cuando estas pruebas puedan utilizarse como pruebas complementarias de ayuda en el establecimiento de una hipótesis diagnóstica, deben cumplir para su prescripción con una serie de criterios, que no sólo están consensuados por guías internacionales, sino que su aplicación también puede suponer un importante ahorro, tanto en el esfuerzo diagnóstico como en medios materiales.
Cariotipo convencionalEl cariotipo convencional ha sido durante años la única herramienta para abordar las causas genéticas del retraso mental o del desarrollo y permite realizar un rastreo del genoma completo, pero está limitado a la detección de anomalías superiores en tamaño a 5Mb.
Los estudios cromosómicos son procedimientos sistemáticos de laboratorio importantes en la evaluación del retraso mental o del retraso del desarrollo, cuya indicación debe aplicarse sin importar la presencia o la ausencia de caracteres dismórficos, talla baja, anomalías congénitas o fenotipo conductual (recordando que el retraso del desarrollo, para la prescripción de pruebas genéticas, debe implicar déficit de aprendizaje y adaptativos “significativos”, no leves) (tabla 2).
Indicaciones para el estudio del cariotipo en pacientes con retraso mental o el desarrollo
|
CI: cociente intelectual.
Estudios importantes basados en la evidencia indican que el total de positivos de la citogenética sistemática es del 3,7 %.
Existen diferentes recomendaciones respecto a la realización en los casos de retraso mental o del desarrollo de estudios cromosómicos constitucionales de alta resolución (resolución superior a 650 bandas o generalmente sobre 850 bandas como recomendación específica)4. A este respecto no parecen estar indicados de forma sistemática, a menos que se requiera investigar una región cromosómica concreta, o exista una historia familiar de una anomalía o fenotipo sindrómico específico, y deben usarse cuando el diagnóstico mediante hibridación in situ (ISH) con sondas de ADN no esté disponible. Dependiendo de la resolución obtenida, la frecuencia de anomalías cromosómicas detectadas puede variar entre un 9 y un 36%5.
Test para descartar el síndrome del cromosoma X frágilEl test para descartar el síndrome del cromosoma X frágil consiste en el estudio molecular de la expansión CGC en el gen FRAXA. Debido al bajo coste de la prueba y a la alta prevalencia en pacientes con retraso mental, se utiliza como prueba complementaria sistemática, aunque siempre debieran tenerse en cuenta algunos aspectos de la clínica (tabla 3).
Indicaciones para la realización de la detección del síndrome del cromosoma X frágil en pacientes con retraso mental o del desarrollo
|
Utilizando los criterios propuestos por el American College of Medical Genetics (ACMG), la tasa de diagnósticos da como resultado una media de aproximadamente un 2,6 % de positivos6.
Síndromes de microdeleciónCuando el paciente presenta características que recuerdan a algún síndrome concreto, y éste está englobado entre los denominados síndromes de microdeleción, las técnicas de hibridación in situ fluorescente (FISH) son particularmente útiles en el diagnóstico de los síndromes de microdeleción, dado que se utilizan sondas específicas de localización de reserva, y su uso debe estar limitado a los casos en los que el fenotipo sugiere un síndrome o una enfermedad específica.
Para muchos de los síndromes de microdeleción existen criterios clínicos estandarizados que son de gran utilidad a la hora de realizar prescripciones de estudios genéticos dirigidos en estos casos. Por ejemplo, en el síndrome de Angelman, entre los criterios para solicitar estudios genéticos dirigidos para la confirmación se incluyen una historia de desarrollo y la presencia de las características clínicas incluidas en los grupos A y B que se resumen en la tabla 4.
Desarrollo y características clínicas en el síndrome de Angelman*
Desarrollo en el síndrome de Angelman
|
EEG: electroencefalograma; RM: resonancia magnética; TC: tomografía computarizada.
Otras condiciones clínicas que pueden ser similares a las características clínicas del síndrome de Angelman, en especial durante la infancia, son el síndrome de Rett, la deleción terminal 22q13.3, el síndrome de Mowat-Wilson, el síndrome de retraso mental ligado al cromosoma X con alfatalasemia (ATR-X), el síndrome de Lennox-Gastaut, la encefalopatía estática con retraso mental, el autismo infantil y la parálisis cerebral no específica, entre otras7.
En el caso del síndrome de Prader-Willi, el análisis FISH es válido clínicamente para la detección de la deleción 15q11-q13, que se encuentra en el 70 % de los casos con fenotipo típico de Prader-Willi. Cuando los estudios de ISH son normales, deben indicarse estudios de metilación para descartar tanto la unidisomía parental (en el 28% de los casos) como mutaciones en el centro de imprinting. Los criterios para la indicación del test genético se resumen en la tabla 58.
Criterios sugeridos para realizar un test genético de síndrome de Prader-Willi
| De 0 a 2 años | Hipotonía con dificultades en la succión |
| De 2 a 6 años | Hipotonía con historia de dificultades en la succión |
| Retraso global del desarrollo | |
| De 6 a 12 años | Hipotonía con historia de dificultades en la succión (la hipotonía puede persistir) |
| Retraso global del desarrollo | |
| Excesivo apetito (hiperfagia, obsesión con la comida) con obesidad central si no se controla | |
| Mayor de 13 años | Discapacidad cognitiva, habitualmente retraso mental moderado |
| Excesivo apetito (hiperfagia, obsesión con la comida) con obesidad central si no se controla | |
| Hipogonadismo hipotalámico y/o fenotipo conductual (obsesividad-compulsividad y cambios de temperamento) |
Dado que el retraso mental o el retraso del desarrollo idiopático se produce con una proporción significativa de casos moderados y leves (un 40 y un 45 %, respectivamente), y dado que muchos de estos casos no presentan un fenotipo característico de un síndrome de microdeleción, el abordaje de confirmación genética no siempre es posible utilizando FISH con una sonda específica de locus o región cromosómica (tabla 6).
Indicaciones para el estudio de los síndromes de microdeleción en pacientes con retraso mental o retraso del desarrollo
|
CI: cociente intelectual.
Recientemente el estudio de las regiones subteloméricas de los cromosomas parece estar siendo de particular utilidad en el ámbito del análisis de las causas genéticas del retraso mental o del desarrollo. Aunque existen diferencias debidas a los distintos criterios de inclusión en los estudios, se estima que aproximadamente un 5–10 % de los casos podrían llegar a ser diagnosticados mediante este tipo de estudios9–11. Técnicamente se estudian mediante amplificación múltiple dependiente de ligamiento (MLPA) subtelomérico o FISH subtelomérico. Ambas técnicas tienen una resolución mucho mayor incluso que el estudio citogenético de alta resolución, pero no permiten el análisis de todo el genoma, sino que están limita das a regiones muy concretas y terminales de los cromosomas. Las técnicas FISH y MLPA son diez veces más sensibles a este respecto12,13.
El grado de retraso del desarrollo o mental es el principal factor predictor en este tipo de estudios. Los retrasos del desarrollo o retrasos mentales moderados o graves asociados con dismorfia facial, anomalías físicas menores en las manos o en los pies, baja talla y/o microcefalia están más probablemente asociados con anomalías subcromosómicas. La presencia de una historia familiar positiva de retraso del desarrollo/retraso mental o fenotipo peculiar incrementa la probabilidad de ser portadores este tipo de reordenamientos. Los resultados de los estudios publicados hasta el momento indican que podrían ser responsables del 10 % de los casos de retraso mental moderado-grave idiopático.
Recientemente se han propuesto una serie de criterios que pueden incluirse como buenos indicadores para el cribado subtelomérico en pacientes con retraso del desarrollo o retraso mental moderado o grave con cariotipo normal: historia familiar positiva de retraso del desarrollo o mental, y/o pérdidas fetales, historia prenatal de retraso de crecimiento, dos o más características dismórficas faciales y una o más características dismórficas no faciales y/o anomalías congénitas14. En la tabla 7 se detallan estos indicadores y puede verse cómo se aplicaría un baremo numérico en el cual el corte para la que la indicación de la prueba sea consistente sería de 3 puntos o más.
Indicaciones para el estudio de deleciones subteloméricas (FISH o MLPA subtelomérico)
| En pacientes con retraso mental moderado o grave si se consigue al menos una puntuación de 3 o más según los siguientes criterios: | ||
| A. Historia familiar de retraso mental y/o pérdidas fetales | ||
| – Herencia mendeliana | 1 | |
| – Herencia no mendeliana o fenotipos discordantes | 2 | |
| B. Historia prenatal | ||
| – Retraso del crecimiento | 2 | |
| – Microcefalia | 1 | |
| C. Anomalías de crecimiento posnatales | ||
| – Talla baja | 1 | Máx. 2 |
| – Macrocefalia | 1 | |
| – Talla alta | 1 | |
| D. Dismorfias faciales (deben concurrir al menos dos signos) | ||
| – Hipertelorismo notable | 1 | Máx. 2 |
| – Anomalías nasales | 1 | |
| – Anomalías de los pabellones auditivos | 1 | |
| E. Defectos congénitos estructurales | ||
| – Anomalías en manos y pies | 1 | Máx. 2 |
| – Anomalías cardíacas | 1 | |
| – Hipospadias ± testes internos | 1 | |
Los microarrays de hibridación genómica comparada (CGH-arrays) son un nuevo método alternativo de rastreo de pequeñas pérdidas y/o ganancias de material genético en el paciente que permite la identificación de alteraciones inferiores a 1Mb. Todavía tienen un alto coste y se están llevando a cabo estudios para establecer su utilidad diagnóstica.
Estas limitaciones pueden paliarse con la utilización de CGH-arrays que sirven para la detección de reordenamientos que afectan a regiones de menos de 1Mb, y permiten, además, un rastreo de todo el genoma en una única reacción de hibridación. Se trata de una técnica basada en la combinación de la metodología de los microarrays con la hibridación genómica comparada (CGH). La resolución de esta técnica viene determinada por la distancia entre los segmentos del ADN que se usan en el microarray como dianas, y por la longitud de éstos, que puede ser de 3Mb, 1Mb, o incluso menor de 100kb.
Los resultados de los estudios publicados hasta el momento indican que con el estudio de arrays de genoma completo podrían diagnosticarse un 10 % más de casos de retraso mental o del desarrollo que con la combinación de estudios de citogenética convencional (cariotipo) y/o FISH (síndromes de microdeleción, estudios de deleciones subteloméricas).
Las recomendaciones actuales del ACMG al respecto15 indican que el uso de los CGH-arrays debe limitarse por el momento, en lo relativo al retraso mental o del desarrollo a su uso, junto con los test citogenéticos habituales (citogenética convencional, FISH y/o MLPA) a la evaluación de pacientes que presentan, además de retraso mental, anomalías congénitas. Los factores que deben considerarse en su aplicación son los siguientes: las limitaciones de financiación, la posible ambigüedad de los resultados y la disponibilidad de muestras de los progenitores.
Estudios de genética molecularCuando se dispone de ellos, los estudios de genética molecular permiten dirigir el estudio hacia una enfermedad muy concreta. Algunos estudios de diagnóstico molecular, como el estudio del síndrome del cromosoma X frágil (genes FraxA) se utilizan como pruebas complementarias sistemáticas. Sin embargo, en algún caso, la interpretación del tipo de herencia, junto con la clínica del paciente, pueden permitir establecer un juicio diagnóstico que permita indicar de forma contundente un test genético para excluir mutaciones en un gen específico implicado en el retraso mental o del desarrollo (p. ej., análisis molecular de LIS1 en el síndrome de Dandy-Walker, o mutaciones RSK2 en el síndrome de Coffin-Lowry). En estos casos, el estudio debería indicarse siempre y cuando la clínica del paciente cumpla criterios consistentes para la realización de estos estudios.
OTROS ABORDAJES DESDE EL PUNTO DE VISTA GENÉTICOSi la evaluación por parte del médico no conduce a ningún diagnóstico claro y el estudio de las anteriores pruebas de laboratorio es negativo podemos encontrarnos ante dos situaciones en la revaluación del diagnóstico:
- 1.
Si existen antecedentes familiares, la familia cuenta con la estructura adecuada (varios varones afectados en distintas generaciones, al menos 3 varones y dos generaciones; de forma ideal, 7 meiosis), la herencia es compatible con una herencia ligada al cromosoma X, y se puede realizar un estudio de ligamiento con marcadores polimórficos a lo largo del cromosoma X. Este estudio debe continuarse con la selección de los genes en la región ligada, y el análisis de mutaciones de éstos, lo cual resulta extremadamente laborioso. Si los estudios son positivos para una alteración molecular en alguno de los genes candidatos podría afirmarse que se ha llegado a un diagnóstico, y se podrá ofrecer consejo genético y diagnóstico prenatal. Si no es así, el paciente quedaría sin diagnóstico.
- 2.
Cuando el patrón de herencia es autosómico dominante o recesivo no podría realizarse ningún otro estudio, salvo que existiera la sospecha clínica de alguna enfermedad conocida (en cuyo caso podría realizarse un análisis de ligamiento, siendo limitante, sobre todo, la estructura familiar en las enfermedades recesivas).
Sin embargo, pocas veces es posible dirigir el estudio o identificar un patrón de herencia claro, por lo que cabría considerar los siguientes aspectos:
- 1.
En las historias sin antecedentes familiares y, fundamentalmente, si el caso índice es un varón, puede realizarse un estudio mediante MLPA de 14 genes ligados al cromosoma X relacionados con el retraso mental o del desarrollo. Esta técnica no es un estudio de mutaciones, por tanto no descarta los síndromes descritos en la tabla 4, sino que evalúa si se producen duplicaciones o deleciones de estos genes.
- 2.
Tanto en varones como en mujeres podría estar indicado el estudio de mutaciones del gen MECP2 que, además de causar el síndrome de Rett, ocasiona una gran variedad de fenotipos en el varón: encefalopatía neonatal, síndrome de Angelman mimético, PPM-X (retraso mental, psicosis, signos piramidales y macroorquidismo). Se identifican mutaciones en un 6,8 % de los varones con retraso mental ligado al cromosoma X.
En cualquier caso, y aunque se carezca de la posibilidad de diagnóstico genético, siempre debería ofrecerse un consejo genético adecuado a la familia.
En resumen, creemos que la etiología del retraso mental o del desarrollo es, como hemos mencionado, variada. Aunque se estima que las causas genéticas podrían llegar a estar implicadas de forma importante en esta afección, incluso en los casos en los que existen antecedentes familiares, procesos como la herencia multifactorial o la penetrancia genética variable resultan una dificultad añadida a la hora de llegar a una confirmación diagnóstica.
En la evaluación del retraso mental o del desarrollo de origen idiopático los tests genéticos utilizados sitemáticamente hasta el momento son los siguientes: la citogenética convencional (cariotipo), la detección de síndromes de microdeleción (FISH específico de locus) y el diagnóstico de mutaciones del gen FraxA. Sin embargo, los avances en las técnicas de diagnóstico genético (FISH y/o MLPA subtelomérico y CGH-arrays) permiten, cada vez más, acercarnos a tasas de positivos más altas. Conseguir este objetivo supone la utilización de pruebas de diagnóstico genético con criterios consensuados para su indicación. De otro modo, podríamos estar utilizándolas como pruebas de cribado poblacional, cayendo en una sobreutilización de los recursos personales y materiales de los que se dispone.
En el proceso de elaboración de una hipótesis diagnóstica clínica deben evaluarse todos los detalles con una metodología rigurosa, que debe incluir la recopilación de una historia clínica lo más completa y ordenada posible, sin olvidarse de la historia familiar y preconcepcional.
Tanto las pruebas complementarias como las pruebas de confirmación de un diagnóstico genético deben ser solicitadas estableciendo un plan diagnóstico, de forma secuencial y respetando los criterios para su indicación, lo cual permite maximizar la tasa de diagnósticos positivos.
Parece claro, por tanto, que la clave principal es lograr un diagnóstico preciso, al que llegaremos mediante un juicio o hipótesis clínica y la realización de los tests adecuados, tanto complementarios como de confirmación. Para ello, en este trabajo se realiza una recopilación de las indicaciones de consenso, provenientes de diferentes guías publicadas por las sociedades y asociaciones científicas nacionales e internacionales16–18, en relación con las diferentes pruebas genéticas que deben aplicarse en el ámbito del retraso mental o del desarrollo. En la figura 1 también se resumen, en forma de algoritmo, los consensos publicados para el diseño de un plan diagnóstico.
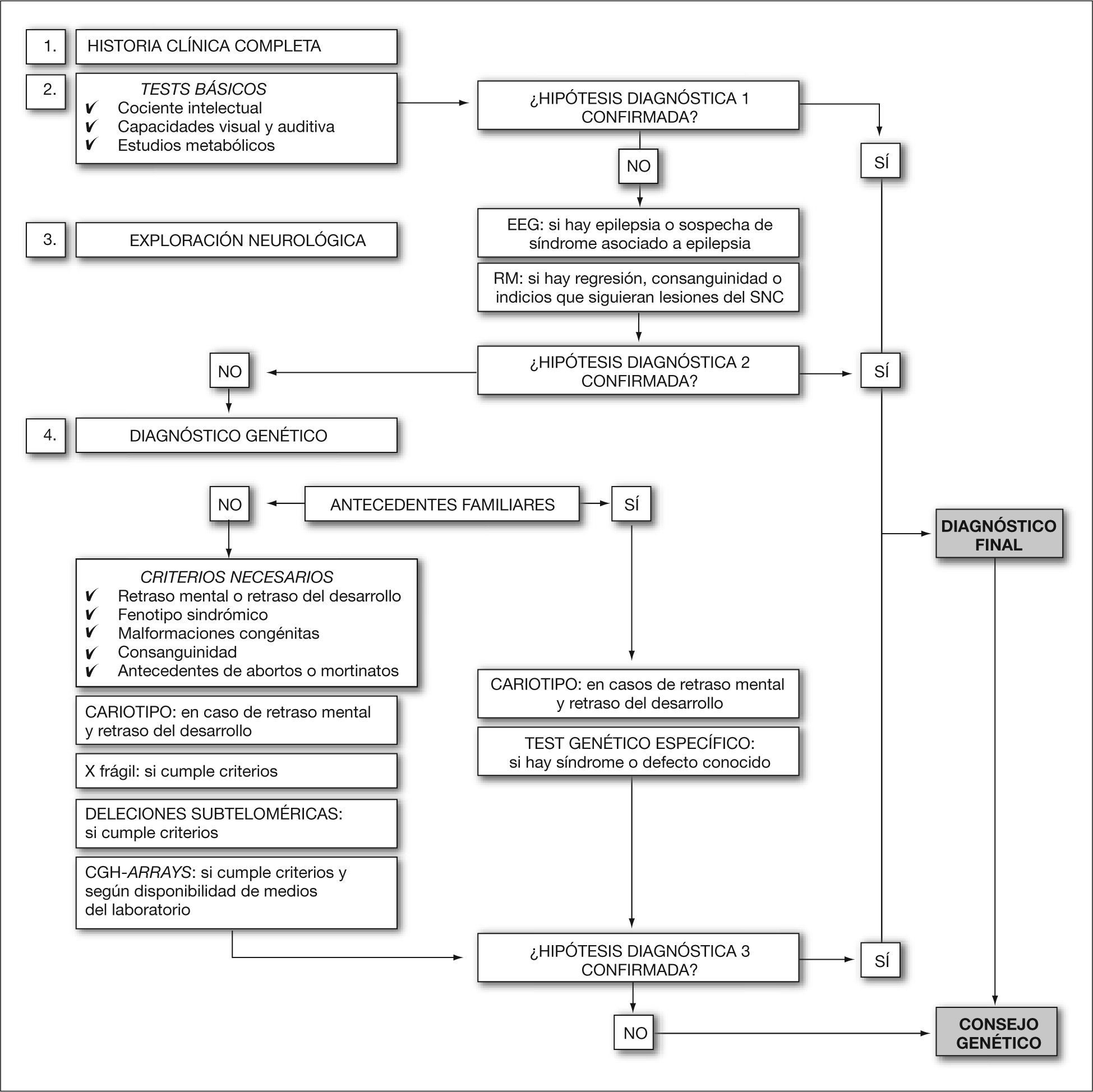
La identificación de la causa, hoy día, pocas veces influye en el tratamiento del paciente afectado, pero permitirá un seguimiento adecuado y un mayor conocimiento sobre su pronóstico. En muchos casos ayudará a conocer el riesgo de recurrencia para futuros embarazos y se podrá proporcionar un adecuado consejo genético al paciente o a su familia, así como la posibilidad de realización de diagnóstico prenatal y detección de portadores en familiares. En este contexto, que forma parte del asesoramiento genético, el acceso a una información completa, comprensible y cercana, es de gran importancia para los padres, ya que puede paliar gran parte de las dificultades psicosociales con las que se encuentran, así como hacerles asimilar sus límites y las pautas adecuadas respecto a los cuidados y responsabilidad educativa de estos pacientes. Lo que hoy conocemos como una integración y educación especial para pacientes con retraso mental o retraso del desarrollo puede que en un futuro difiera en protocolos de actuación psicopedagógica, según el tipo de enfermedad del paciente. Por todo lo que pueda ocurrir en el futuro es por lo que debemos trabajar hoy.
