
Gaucher disease (GD) is caused by deficiency of the enzyme acid beta-glucosidase (GBA).1 The currently accepted classification is: type 1 (non-neuropathic) (GD-1), type 2 (acute neuropathic) (GD-2) and type 3 (chronic neuropathic). Since 1994, enzyme replacement treatment (ERT) has been available in Spain. Due to the limited long-term safety and efficacy experience with ERT, it was of interest to describe the experience in a tertiary pediatric hospital in which, this treatment started being used as soon as it was available.
The evolution of 7 patients with GD (5 GD-1 and 2 GD-2) treated within the past 20 years in Hospital Infantil La Fe (Valencia) was retrospectively described. The diagnosis was confirmed by glucocerebrosidase activity in leukocytes (GAL) and by mutations in the GBA gene. At diagnosis, and yearly thereafter, weight, height, BMI, hepatomegaly–splenomegaly degree, hemogram, hemostasis, chitotriosidase, chemokine ligand 18 (CCL18) and spine densitometry were assessed. Abdominal magnetic resonance imaging (MRI) was performed to assess liver and spleen volumes, calculating the multiples of their normal volumes (MNV).2 In patients with bone involvement, MRI of long bones was performed. ERT was administered by iv infusion for 2h, every 2 weeks at a 60UI/kg dose in patients with GD-1.
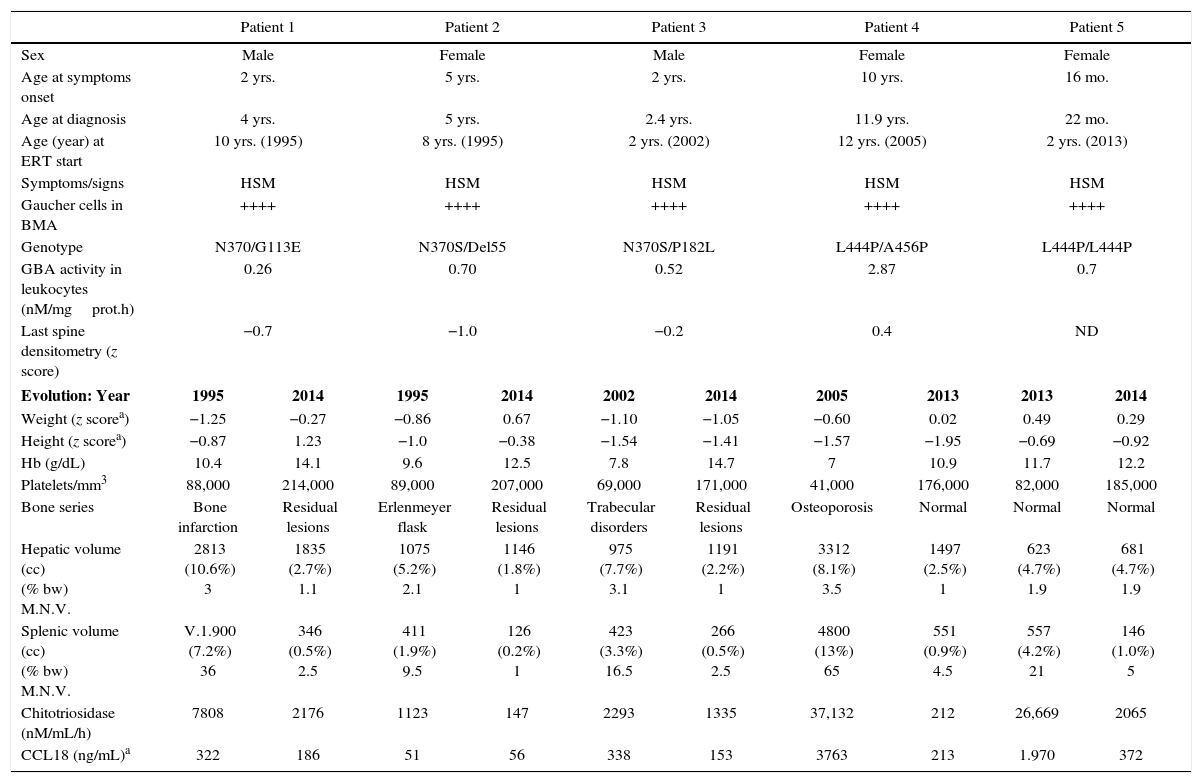
Table 1 shows evolution of the 5 patients with GD-1. Age at diagnosis ranged between 22 months and 11 years. In every case, bone marrow aspiration showed Gaucher cells. Genetic analysis showed that 3 of the patients carried the mutation N370S in heterozygosis and 2 the mutation L444P. Every patient showed hepatosplenomegaly and anemia. The initial hepatomegaly was 1.9–3.5 times MNV, and splenomegaly was 9–65 times MNV. One patient presented with lesions of bone infarction and two had Erlenmeyer-flask shaped femurs. GAL ranged from 0.26–2.87nM/mgprot.h (lower than 8% of control GAL in every case). Patients 1 and 2 received treatment with alglucerase from 1995 to 1998 and with imiglucerase thereafter. Every patient improved his/her weight and height, hemoglobin and platelets count. ERT reduced the hepatic volume up to 3 times and that of the spleen 4–15 times. These improvements occurred mainly during the first 12–24 months of treatment. The patients did not need blood transfusions. In 2009, due to the 6-month shortage of imiglucerase,3 2 patients started taking miglustat. Both patients showed an increase in chitotriosidase and CCL18; both biomarkers returned to normal values when ERT was reintroduced at previous doses. These patients did not show hematologic or bone involvement and no modification of the hepatosplenomegalies. In 4 GD-1 patients, ERT was initiated at an age earlier than 10 years (in 2 of them at two years). Two patients have received ERT for 20 years and one for 13 years. In every case no adverse events in relation to ERT and the therapeutic objectives have been achieved and sustained.4 The mutation N370S protects against neurologic involvement, while mutation L444P, which is present in 77% of alleles of neuropathic GD, increases the risk of this form of disease presentation. However, the coexistence of determined polymorphisms may modify this risk.5 Patient 4, neurologically asymptomatic, showed MRI hyperintense lesions in the periventricular white matter, which have remained stable. Likewise, patient 5, homozygous for L444P, showed normal brain MRI and neurologic exploration and his response to ERT was good.
Baseline characteristics and evolution of patients with GD type 1.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Male | Female | Female | |||||
| Age at symptoms onset | 2 yrs. | 5 yrs. | 2 yrs. | 10 yrs. | 16 mo. | |||||
| Age at diagnosis | 4 yrs. | 5 yrs. | 2.4 yrs. | 11.9 yrs. | 22 mo. | |||||
| Age (year) at ERT start | 10 yrs. (1995) | 8 yrs. (1995) | 2 yrs. (2002) | 12 yrs. (2005) | 2 yrs. (2013) | |||||
| Symptoms/signs | HSM | HSM | HSM | HSM | HSM | |||||
| Gaucher cells in BMA | ++++ | ++++ | ++++ | ++++ | ++++ | |||||
| Genotype | N370/G113E | N370S/Del55 | N370S/P182L | L444P/A456P | L444P/L444P | |||||
| GBA activity in leukocytes (nM/mgprot.h) | 0.26 | 0.70 | 0.52 | 2.87 | 0.7 | |||||
| Last spine densitometry (z score) | −0.7 | −1.0 | −0.2 | 0.4 | ND | |||||
| Evolution: Year | 1995 | 2014 | 1995 | 2014 | 2002 | 2014 | 2005 | 2013 | 2013 | 2014 |
| Weight (z scorea) | −1.25 | −0.27 | −0.86 | 0.67 | −1.10 | −1.05 | −0.60 | 0.02 | 0.49 | 0.29 |
| Height (z scorea) | −0.87 | 1.23 | −1.0 | −0.38 | −1.54 | −1.41 | −1.57 | −1.95 | −0.69 | −0.92 |
| Hb (g/dL) | 10.4 | 14.1 | 9.6 | 12.5 | 7.8 | 14.7 | 7 | 10.9 | 11.7 | 12.2 |
| Platelets/mm3 | 88,000 | 214,000 | 89,000 | 207,000 | 69,000 | 171,000 | 41,000 | 176,000 | 82,000 | 185,000 |
| Bone series | Bone infarction | Residual lesions | Erlenmeyer flask | Residual lesions | Trabecular disorders | Residual lesions | Osteoporosis | Normal | Normal | Normal |
| Hepatic volume (cc) (% bw) M.N.V. | 2813 (10.6%) 3 | 1835 (2.7%) 1.1 | 1075 (5.2%) 2.1 | 1146 (1.8%) 1 | 975 (7.7%) 3.1 | 1191 (2.2%) 1 | 3312 (8.1%) 3.5 | 1497 (2.5%) 1 | 623 (4.7%) 1.9 | 681 (4.7%) 1.9 |
| Splenic volume (cc) (% bw) M.N.V. | V.1.900 (7.2%) 36 | 346 (0.5%) 2.5 | 411 (1.9%) 9.5 | 126 (0.2%) 1 | 423 (3.3%) 16.5 | 266 (0.5%) 2.5 | 4800 (13%) 65 | 551 (0.9%) 4.5 | 557 (4.2%) 21 | 146 (1.0%) 5 |
| Chitotriosidase (nM/mL/h) | 7808 | 2176 | 1123 | 147 | 2293 | 1335 | 37,132 | 212 | 26,669 | 2065 |
| CCL18 (ng/mL)a | 322 | 186 | 51 | 56 | 338 | 153 | 3763 | 213 | 1.970 | 372 |
HSM: hepatosplenomegaly; ND: no data; CCL18: chemokine (C-C motif) ligand 18; BMA: bone marrow aspiration; GBA: glucocerebrosidase; Hb: hemoglobin; % bw: percentage with respect to body weight; M.N.V.: multiple of normal value (normal value: liver: 2.5% of body weight; spleen: 0.2% of body weight).
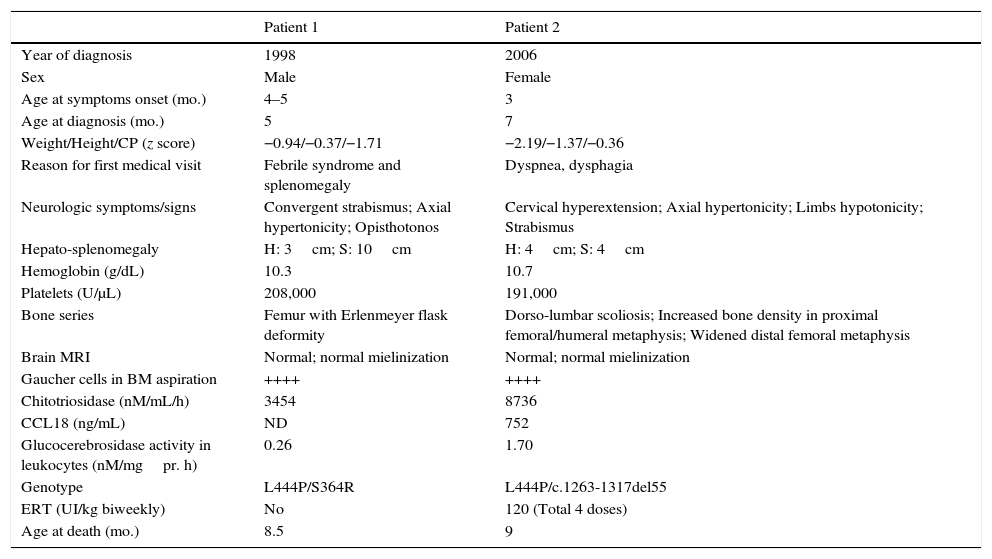
As Table 2 shows, patients with GD-2 showed hepato-splenomegaly, bone disorders and increased chitotriosidase but no major lesions in brain MRI. Both had the mutation L444P in heterozygosis and died before 9 months of life, and one of these patients was treated with 4 doses of ERT at 120UI/kg. It has to be pointed out that normal MRI does not rule out GD-2.
Baseline characteristics of patients with GD type 2.
| Patient 1 | Patient 2 | |
|---|---|---|
| Year of diagnosis | 1998 | 2006 |
| Sex | Male | Female |
| Age at symptoms onset (mo.) | 4–5 | 3 |
| Age at diagnosis (mo.) | 5 | 7 |
| Weight/Height/CP (z score) | −0.94/−0.37/−1.71 | −2.19/−1.37/−0.36 |
| Reason for first medical visit | Febrile syndrome and splenomegaly | Dyspnea, dysphagia |
| Neurologic symptoms/signs | Convergent strabismus; Axial hypertonicity; Opisthotonos | Cervical hyperextension; Axial hypertonicity; Limbs hypotonicity; Strabismus |
| Hepato-splenomegaly | H: 3cm; S: 10cm | H: 4cm; S: 4cm |
| Hemoglobin (g/dL) | 10.3 | 10.7 |
| Platelets (U/μL) | 208,000 | 191,000 |
| Bone series | Femur with Erlenmeyer flask deformity | Dorso-lumbar scoliosis; Increased bone density in proximal femoral/humeral metaphysis; Widened distal femoral metaphysis |
| Brain MRI | Normal; normal mielinization | Normal; normal mielinization |
| Gaucher cells in BM aspiration | ++++ | ++++ |
| Chitotriosidase (nM/mL/h) | 3454 | 8736 |
| CCL18 (ng/mL) | ND | 752 |
| Glucocerebrosidase activity in leukocytes (nM/mgpr. h) | 0.26 | 1.70 |
| Genotype | L444P/S364R | L444P/c.1263-1317del55 |
| ERT (UI/kg biweekly) | No | 120 (Total 4 doses) |
| Age at death (mo.) | 8.5 | 9 |
ERT: enzyme replacement therapy; BM: bone marrow; MRI: magnetic resonance imaging; ND: no data; CP: cephalic perimeter; CCL18: chemokine (C-C motif) ligand 18.
As a summary, during the 20-year period in which ERT has been used for GD patients in our center, we have observed the evolution of patients with different characteristics: (a) three patients with GD-1 with the mutation N370S have evolved well, except for the ERT shortage period that affected 2 patients, showing marked increases of chitotriosidase and CCL18. These markers are more sensitive and earlier than the modifications observed in hematologic and bone parameters. (b) Two patients with GD-1 with the mutation L444P did not present any neurologic symptom/sign during 10- and 2-year periods, respectively, in which they were on ERT treatment. (c) The 2 patients with GD-2 died within 2–3 months after diagnosis, independently of treatment with ERT at high doses. This has been the reason why in the current treatment guidelines, the use of ERT is not recommended in GD-2.6
Please cite this article as: Vitoria Miñana I, Dalmau Serra J. Enfermedad de gaucher: tratamiento enzimático sustitutivo iniciado en la edad pediátrica. Experiencia de 20 años. An Pediatr (Barc). 2016;84:343–346.
