

Autoimmune inflammatory myopathies constitute a broad clinical spectrum and, depending on the involved autoantibodies, can have an unfavourable prognosis with involvement of different organs. Anti-signal recognition particle (SRP) antibodies are present in fewer than 1% of children with these diseases and are associated with a poor prognosis, giving rise to necrotising myopathy with poor response to steroid therapy and multiple system involvement, and a high morbidity and mortality.
We present the case of a girl aged 11 years who reported progressive muscle weakness and myalgia with onset 3 months prior, with inability to climb steps or get up from the floor in the past week. She had no personal or family history of interest. The physical examination evinced significant muscle weakness in the axial muscles of the neck, the pelvic girdle, the shoulder girdle and dorsal and lumbar back, and occasional choking on ingestion of fluids, with a score of 9/52 in the Childhood Myositis Assessment Scale. The patient weighed 52 kg and had a body surface area of 1.51 m2. Blood tests evinced elevation of creatine phosphokinase (CPK, 11 426 U/L [normal range, 26–192]), aldolase (94.2 IU/L; [normal range, 1–7.5]), lactate dehydrogenase (LDH, 1513 U/L [normal range, 120–300]), alanine aminotransferase (ALT, 137 IU/L [normal range, 5–31]) and aspartate aminotransferase (AST, 193 U/L [normal range, 10–31]).
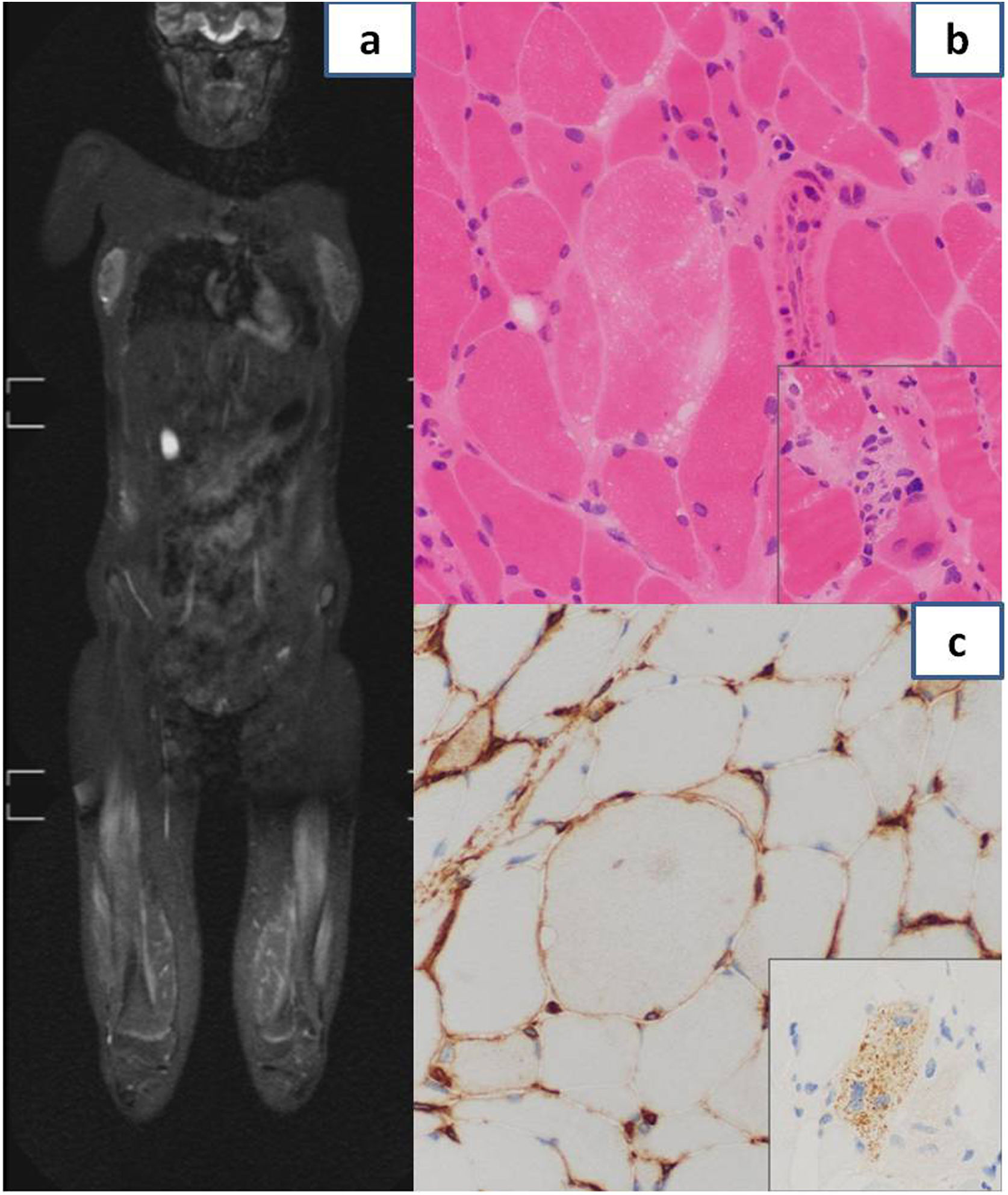
An extensive differential diagnosis was performed during the hospital stay, ruling out infectious, neurologic, metabolic, toxic, endocrinological and oncological causes. The autoimmunity study was positive for anti-SRP-54 and anti-52 kDa Ro/SSA antibodies. Electromyography evinced significant inflammatory myopathy and magnetic resonance imaging muscle changes with a bilateral, symmetrical and multifocal patchy myofascial pattern and oedema predominantly found in the pelvic girdle and the proximal lower extremity muscles (Fig. 1a). Based on the suspicion of immune-mediated inflammatory myositis, high-dose systemic steroid therapy was initiated, which did not achieve a clinical response or, initially, a change in laboratory markers (intravenous boluses of methylprednisolone at 125 mg/day for 5 days followed by prednisone at a dose of 60 mg/day). The muscle biopsy showed myopathic changes with some necrotic and some regenerating fibres and little inflammation. The immunohistochemical analysis revealed focal sarcolemmal HLA class I expression and diffuse expression in the cytoplasm of p62 forming aggregates in scattered fibres, all features compatible with immune-mediated necrotising myositis (Fig. 1b and c). On account of the possibility of systemic involvement, the patient underwent an extensive evaluation that included: cardiological assessment with echocardiography, which evinced adequate biventricular function and absence of valve insufficiency, spirometry and lung diffusion testing evincing adequate lung function, and assessment of swallowing with an upper gastrointestinal series in which the video fluoroscopy findings were normal. Given the lack of response to steroid therapy, intravenous immunoglobulin (1 g/kg), methotrexate (6.6 mg/m2/week: 10 mg) and rituximab (375 mg/m2; first dose: 500 mg with a second dose 15 days later) were added, in addition to intensive physical therapy, which achieved a progressive decrease of muscle enzymes. At present, 2 years after diagnosis, the patient receives methotrexate weekly (5 mg) and intravenous immunoglobulin monthly (1 g/kg), and remains stable in terms of both clinical and laboratory features. The patient has not developed additional symptoms, does not suffer from dysphagia and the cardiovascular and pulmonary assessments show no complications, and her muscular strength has improved significantly, allowing her to be independent in her activities of daily living (Childhood Myositis Assessment Scale 45/52).
 Magnetic resonance imaging showing muscle changes with bilateral, symmetrical and multifocal patchy myofascial pattern, with significant involvement of the quadriceps, hamstring and adductor muscles. (b) Histological examination of muscle biopsy specimen: variation in size of muscle fibres and a necrotic fibre in centre of image without significant inflammation. The inset shows an image characteristic of myophagocytosis. (c) Histological examination of muscle biopsy specimen: focal sarcolemmal HLA class I expression. The inset shows diffuse expression of p62, forming aggregates, in the cytoplasm of a muscle fibre.")
(a) Magnetic resonance imaging showing muscle changes with bilateral, symmetrical and multifocal patchy myofascial pattern, with significant involvement of the quadriceps, hamstring and adductor muscles. (b) Histological examination of muscle biopsy specimen: variation in size of muscle fibres and a necrotic fibre in centre of image without significant inflammation. The inset shows an image characteristic of myophagocytosis. (c) Histological examination of muscle biopsy specimen: focal sarcolemmal HLA class I expression. The inset shows diffuse expression of p62, forming aggregates, in the cytoplasm of a muscle fibre.
When a patient presents with muscle weakness and hyperCKaemia, the differential diagnosis must be broad, including infectious, neurologic, metabolic, toxic, endocrinological, oncological and autoimmune aetiologies. The autoimmune screen can provide information regarding the prognosis of the patient and the changes found in the muscle biopsy examination. The inflammatory myopathies with the least favourable prognoses are necrotising myopathies associated with anti-SRP and anti-HMGCR antibodies.
At present, there are only 40 reported cases of paediatric patients with anti-SRP immune-mediated necrotising myositis in the scientific literature (PubMed search through July 2023). Table 1 presents the clinical characteristics, treatment and outcomes of these patients.
Review of the literature on paediatric cases of anti-SRP immune-mediated necrotising myositis.
| Article (authors and year of publication) | Case (sex, age, race/ethnicity) | Presentation at onset | Laboratory findings | Symptoms | Treatment | Outcome |
|---|---|---|---|---|---|---|
| Rider et al, 1994 | Female, 10 years, Caucasian | Abdominal pain, vomiting, weight loss and weakness | CK 8316 IU/LAldolase 89 IU/L | Cutaneous exanthemaDilated periungual capillariesArthritis in knee and anklesMild pulmonary involvement | Steroid therapyMethotrexateIVIG (1 g/kg) | Clinical improvement |
| Suzuki et al, 2008 | Male, 11 years, Asian | Weakness of trunk and extremities with scapulohumeral atrophy | CK 4180 IU/L | Dysphagia | Steroid therapy | Clinical improvement |
| Rouster-Stevens et al, 2008 (3 cases) | Female, 16 years, African American | Upper arm and neck flexor weakness, wrist arthritis and Raynaud disease | CK 22 155 IU/L | Interstitial pulmonary involvementLVH | Steroid therapyMethotrexateHydroxychloroquineCyclophosphamideTacrolimusMycophenolateInfliximab | Ovarian failureHaemolytic uraemic syndromeNew post-infectious episodeSevere mobility impairment |
| Female, 14 years, African American | Weakness of upper and lower extremities, wrist arthritis following infectious disease | CK 22 857 IU/L | Interstitial pulmonary involvement | Steroid therapyMethotrexateMycophenolateCiclosporinInfliximabIVIG IV (1 g/kg) | Recurrence with new infectionSevere mobility impairment | |
| Female, 11 years, African American | Proximal limb weakness, arthritis and Raynaud disease following a viral respiratory infection | CK 33 000 IU/L | DysphagiaLVH | Steroid therapyMethotrexateMycophenolateCiclosporinInfliximabAzathioprineIVIG (1 g/kg) | Mild mobility impairment | |
| Takada et al, 2009 | Female, 17 years, Asian | Skin rash | CK 6543 IU/L | – | Steroid therapyCiclosporin | Clinical improvement |
| Suzuki et al, 2011 (2 cases) | Female, 5 years, Asian | Frequent fallsProximal muscle weakness in extremities and muscle atrophy | CK 4629 IU/L | – | Steroid therapy | Severe mobility impairment |
| Female, 9 years, Asian | Seven months of extremity and trunk weakness, frequent falls | CK 2467 IU/L | – | Steroid therapyMethotrexateCyclophosphamideTacrolimus | Severe mobility impairment | |
| Kawabata et al, 2012 | Female, 15 years, Asian | Asymptomatic CK elevation following infectious disease. At 1 month, proximal extremity weakness and progressive asthenia | CK 20 375 IU/LAldolase 236 IU/L | Dysphagia | Steroid therapyCyclophosphamideAzathioprinePlasmapheresis | Mild mobility impairment |
| Luca et al, 2012 | Female, 12 years | Proximal weakness, alopecia, dysphagia and Raynaud disease in the past month | CK 8825 IU/L | Pulmonary involvement | Steroid therapyMethotrexateAzathioprineIVIGRituximabLeflunomide | Marked improvement |
| Rider et al, 2013 (6 cases) | 4 female, 2 male 11.6−16.1 years | Two with insidious course, 1 with slow progression and 3 with acute onset of muscle weakness | CK 9111−22 857 IU/L | Dysphagia 50%Pulmonary involvement 83%-Cardiac involvement 50%-Cutaneous involvement 66% | – | – |
| Monomura et al, 2014 | Female, 15 years, Asian | Weakness of upper extremities following infectious disease, with subsequent development of weakness in lower extremities and asthenia | CK 20 375 IU/LAldolase 263 IU/L | Dysphagia | Steroid therapyPlasmapheresisCyclophosphamideAzathioprine | Marked improvement at 1 year. Able to jog |
| Suzuki et al, 2015 (5 cases) | 8 patients | – | – | – | – | – |
| Kobayashi et al, 2016 | Female, 8 years, Asian | Muscle pain and weakness of 2 years’ durationAcute progression following influenza infection | CK 5896 IU/LAldolase 63.9 IU/L | – | Steroid therapyMethotrexateIVIGTacrolimus | Improvement of weakness |
| Zao et al, 2017 (3 cases) | Female, 4 years | Weakness in lower and upper extremities of 6 months’ duration. Myalgia | CK 4020 IU/L | – | IVIG (0.4 mg/kg)Steroid therapy | After 12 months, able to stand without assistanceMarked improvement |
| Female, 11 years | Proximal weakness of upper and lower extremities, rapid progression in 2 meses | CK 4660 IU/L | – | IVIG (0.4 mg/kg)Steroid therapy | At 18 months, able to walk without assistance, marked improvement | |
| Female, 12 years | Weakness of lower extremities with onset 2 years prior and rapid progression to the upper extremities | CK 13 265 IU/L | Dysphagia | IVIG (0.4 mg/kg)Steroid therapy | Marked improvement | |
| Binns et al, 2017 (3 cases) | Female, 14 years | 4 months of proximal weakness, swollen ankles, myalgia and headache | CK 23 111 IU/L | DysphagiaPulmonary involvement | Steroid therapyMethotrexateRituximabIVIG (2 g/kg) | Pneumonitis due to CMVMarked improvement |
| Female, 13 | Four weeks of proximal weakness, swollen eyelids, myalgia and dyspnoea | CK 25 937 IU/L | Pulmonary involvement | Steroid therapyMethotrexateRituximabCyclophosphamideIVIG (2 g/kg) | Marked improvement | |
| Female, 11 | Proximal weakness, myalgia and arthralgia of one week’s duration following a respiratory infection | CK 19 808 IU/L | Pulmonary involvement Bronchoaspiration | Steroid therapyMethotrexateRituximabCyclophosphamideIVIG (2 g/kg) | Marked improvement | |
| Kishi et al, 2017 (8 cases) | 10.7−16 years | – | – | – | – | – |
| Yi et al, 2021 | Female, 8 years, Asian | Acute muscle weakness | CK 28 819 IU/L | Dysphagia | Steroid therapyRituximab | Marked improvement |
| Della Marina et al, 2021 | Female, 8 years | Muscle weakness of 8 months’ duration | CK 10 710 IU/LAldolase 127 IU/L | DysphagiaPulmonary involvement | Steroid therapyMethotrexateIVIG (2 g/kg)Rituximab | Improvement, but with persistence of Gower’s sign and difficulty climbing stairsAvascular femoral necrosis |
| Toledo del Castillo et al, 2024 | Female, 10 years, Caucasian | Three months of proximal muscle weakness in extremities and trunk | CK 11 426 IU/LAldolase 94.2 IU/L | Dysphagia | Steroid therapyMethotrexateRituximabIVIG | Marked improvement |
CK, creatine kinase; CMV, cytomegalovirus; IVIG, intravenous immunoglobulin; LVH, left ventricular hypertrophy.
It is important to differentiate this disease from other idiopathic inflammatory myopathies in children, given its poor response to conventional treatment and systemic involvement associated with significant morbidity and mortality and frequent relapses. This disease should be suspected in patients with significant proximal weakness in absence or with minimal cutaneous lesions and CK values greater than 10 000 IU/L (20–50 times the upper limit of normal).1
The cases published in the literature are consistent in the poor response to methotrexate/cyclophosphamide associated with steroids. The most recent articles have reported improved outcomes with monthly treatment with intravenous immunoglobulin at a dose of 1–2 g/kg combined with steroids and methotrexate, in addition to rituximab and intensive physical therapy. The use of B cell depletion therapies, chiefly rituximab, is achieving good results in adult patients with anti-SRP immune-mediated necrotising myositis, and is recommended in the current literature as part of an early intensive treatment approach for this disease.2–5 Other drugs used in the previous literature include azathioprine, mycophenolate mofetil, tacrolimus, ciclosporin and abatacept, although the data on their use are still scarce.2
The prognosis of the disease is poorer at early ages, when there is a rapid replacement of affected muscle fibres by fat, so aggressive initial treatments seem to be the best options to prevent disability.3
In conclusion, necrotising myositis should be suspected in patients presenting with proximal muscle weakness that progresses rapidly and with significant elevation of CK in order to make an early diagnosis and start intensive treatment from the early stages, given the lack of response to conventional treatment and the poor prognosis in cases with onset at an early age. Magnetic resonance imaging and, above all, muscle biopsy are useful in severe and refractory cases not only to guide the diagnosis but also due to their prognostic value.

