







Incontinentia pigmenti is a rare genodermatosis of inheritance linked to the X chromosome that affects tissues derived from ectoderm. The aim of the study is to review, as completely as possible, the cases diagnosed in paediatric patients in two hospitals.
Material and methodsA retrospective cross-sectional study was carried out, using the clinical, analytical, radiological, and genetic data of paediatric patients diagnosed with incontinentia pigmenti from 2004 to 2018. The data collected were analysed and evaluated at a multidisciplinary level.
ResultsA total of thirteen patients diagnosed with incontinentia pigmenti were included in the study. All of them were female. A genetic study was performed on 11 patients, which confirmed findings compatible with incontinentia pigmenti in 10 of them. Extracutaneous involvement associated with the disease was observed at neurological level (radiological findings in 6 cases, and clinical expression in 3 of them), ophthalmological level (4 cases), dental level (7 cases), and haematological level (4 cases).
ConclusionsA presentation is given of the most complete study published so far of incontinentia pigmenti in Spain. In this study, the results of the disease manifestations were similar to the largest case series published internationally, which reinforces the importance of a multidisciplinary study and follow-up.
La incontinencia pigmenti es una genodermatosis poco frecuente, de herencia ligada al cromosoma X, que afecta a tejidos derivados del ectodermo. Nuestro objetivo es revisar de la forma más completa posible los casos diagnosticados en edad pediátrica en dos hospitales.
Material y métodosSe ha realizado un estudio transversal retrospectivo, recogiéndose datos clínicos, analíticos, radiológicos y genéticos valorados a nivel multidisciplinar de pacientes diagnosticados en la edad pediátrica de incontinencia pigmenti desde el año 2004 al 2018.
ResultadosSe incluyeron 13 pacientes diagnosticados de incontinencia pigmenti, todas de sexo femenino. Se realizó estudio genético en 11 de las 13, confirmándose alteraciones compatibles en 10 de ellas. Se observó afectación extracutánea relacionada con la enfermedad a nivel neurológico (con alteraciones radiológicas en 6 casos y expresión clínica en 3 de ellas), oftalmológico (4 casos), odontológico (7 casos) y hematológico (4 casos).
ConclusionesPresentamos el estudio más completo publicado hasta ahora de incontinencia pigmenti en España. Los resultados del estudio de las manifestaciones de la enfermedad fueron similares a las series de casos más amplias publicadas a nivel internacional y refuerzan la importancia de un estudio y seguimiento multidisciplinar.
Incontinentia pigmenti (IP) is an infrequent genodermatosis with an X-linked dominant pattern of inheritance and a penetrance of 100%. It affects tissues that develop from the endoderm, causing abnormalities in the skin, hair, teeth, eyes and nervous system.1
It was first described in a patient by Garrod in 1906, and its incidence is estimated at approximately 1 in 40000–50000 births, although the exact prevalence is not known, as it remains an underdiagnosed disease.1,2
The onset usually manifests at the level of the skin in the first 2 weeks post birth, and more than 95% of cases occur in female patients.1 There are four cutaneous stages of IP: stage 1 (vesicular); stage 2 (hyperkeratotic); stage 3 (hyperpigmented); and stage 4 (hypopigmented).3 The timing and duration of each stage vary between individuals, and some stages may overlap or even not occur in some cases.4
The mutation responsible for the disease is in the IKBKG gene (previously known as NEMO), located on locus Xq28. This gene encodes a protein (IKK-gamma or NEMO) that activates transcription factor nuclear-kappa B (NF-kB), which in turn regulates the immune response, the inflammatory cascade and mechanisms of cell apoptosis. In 80–90% of cases, the mutation involves deletion of exons 4–10 of the IKBKG gene, while the rest are due to other genetic changes (missense, nonsense or frameshift mutations, microdeletions, etc.) identified by means of DNA sequencing.2,5
This syndrome is lethal in the male sex except in special situations, such as mosaicism, Klinefelter syndrome (XXY karyotype) or hypomorphic mutations in the IKBKG gene.6
In 1993, Landy and Donnai were the first to propose a series of diagnostic criteria for IP, which were later updated by Minić et al. with the inclusion of molecular genetic testing (Table 1).7,8
Landy and Donnai diagnostic criteria updated by Minić et al.7
| Major criteria | Minor criteria | Criteria to confirm diagnosis |
|---|---|---|
| Typical IP skin stages distributed along Blaschko's lines:• (Stage 1) Vesiculobullous• (Stage 2) Verrucous• (Stage 3) Hyperpigmented• (Stage 4) Atrophic/hypopigmented | Central nervous system/neurologic anomalies: seizures, spastic paralysis, psychomotor retardation, intellectual disability, microcephaly, neurologic abnormalities/CNS anomalies: seizures, spastic paralysis, psychomotor delay, intellectual disability, microcephaly, cerebral/cerebellar atrophy, microgyria/polymicrogyria, hypoplasia of corpus callosum, abnormalities of the basal ganglia, leukomalacia periventricular, hydrocephalus, porencephaly, ischaemic stroke, diffuse haemorrhagic necrosis, encephalomyelitisEye anomalies: vision impairment, retinopathy, retinal detachment, vascular abnormalities in retina, hyper/hypopigmented retina, optic nerve atrophy, macular hypoplasia, retrolental fibroplasia, cataracts, microphthalmia, strabismus, nystagmusDental anomalies: delay in tooth eruption, anodontia/hypodontia, microdontia, dental dystrophy, abnormalities of tooth morphology (conical teeth), impaction, diastema, malocclusionPalate anomalies: high-arched palate altoBreast anomalies: supernumerary nippleHair abnormalities (hair, eyebrows, eyelashes): alopecia, hypertrichosisAbnormal nails: onychodystrophy, yellowish discoloration, horizontal or vertical ridgesMiscarriage of male foetusesTypical pathological and histological features in skin | No evidence of IP in first-degree female relative:If molecular testing is not available, 2 or more major criteria or 1 major criterion and at least 1 minor criteria are needed to confirm the diagnosisMutation in IKBKG in addition to any major or minor criteria confirms the diagnosisEvidence of IP in first-degree female relative:Any single major or at least 2 minor criteriaIn all cases, eosinophilia and skewed X chromosome inactivation support the diagnosis |
Due to the general ignorance of this disease, many mild cases are unnoticed and affected individuals reach adulthood without being diagnosed, which facilitates transmission to the disease to the offspring. In addition, in some cases a diagnosis is made but screening and follow-up of potential associated cutaneous and non-cutaneous abnormalities is not performed. In Spain, other authors have published case series of IP, but usually including fewer patients and with a less thorough evaluation of the disease.4,6,9–11
The primary objective of our study was to provide the most detailed possible review of the clinical manifestations of cases of IP diagnosed during childhood in 2 Spanish hospitals and to compare these data with the findings published in the international literature. Our aim was to increase the level of clinical suspicion of health care providers to achieve earlier diagnosis of the disease and promote the coordination of a multidisciplinary follow-up plan for these patients.
Materials and methodWe included patients that received a diagnosis of IP during childhood based on genetic testing and/or the clinical criteria modified by Minić et al.7 in the Department of Dermatology of the Hospital Costa del Sol (Marbella) or the Hospital Regional Universitario Materno Infantil (Malaga) between 2004 and 2018. We conducted a retrospective cross-sectional study with collection of clinical, laboratory, radiological and genetic data for each patient from the time of the initial suspicion of IP to the latest follow-up appointment (December 2018): age at onset of typical skin lesions and their evolution, other potential dermatological abnormalities in skin, hair, teeth or nails, complete blood count, molecular genetic tests, histological findings (skin biopsy), neuroimaging tests and neurologic manifestations.
Clinical suspicion of IP was always based on the presence of skin lesions, which were the initial symptom and developed in the neonatal period in every case. This led to histologic examination and genetic testing in most cases, with the diagnosis of IP made based on the criteria mentioned above.7
The study was designed and coordinated by the Departments of Dermatology and Paediatric Neurology with involvement of outpatient and inpatient levels of care, and patients underwent a multidisciplinary evaluation (neonatology, dermatology, paediatric neurology, radiology, ophthalmology and genetics). We received informed consent from the parents prior to the performance of procedures and the collection of data for the study.
ResultsWe included a total of 13 patients with an IP diagnosis, all of them female. The clinical follow-up of all patients lasted through December 2018, except for patients 3 and 6, who were lost to follow-up.
Eight of the patients underwent evaluation in the Department of Dermatology in the first 30 days post birth, and the remaining 5 before age 1 year. All patients experienced the onset of cutaneous manifestations during the neonatal period. All had onset with vesicular lesions along the Blaschko's lines, overlapping in 2 cases with lesions in the hyperpigmented stage (case 3) and verrucous stage (case 9).
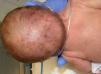
The evolution of skin lesions was varied, with stages alternating and overlapping in time, and in some cases without manifestation of some stage (Fig. 1).
Patients that exhibited more severe inflammatory activity received topical treatment of the cutaneous lesions consisting of corticosteroids in 4 patients (cases 1, 2, 9 and 10) and steroids combined with fusidic acid in 1 patient (case 5), all of who exhibited a good response (Table 2).
Dermatologic manifestations in the case series.
| Case | Age at onset of skin lesions | First assessment in dermatology | Onset of cutaneous manifestations | Evolution of cutaneous manifestations | Treatment of cutaneous lesions | Other anomalies in skin/common integument | Genetic testing (age performed) | Skin biopsy (age performed) |
|---|---|---|---|---|---|---|---|---|
| 1 | First 24h | Neonatal period | Vesicular stage (vesicles in upper and lower limbs and torso along the lines of Blaschko) | 1) Verrucous stage2) Atrophic stage | Topical corticosteroid | Focal infantile superficial haemangioma | 2 months: deletion in exons 4–10 of IKBKG gene | 6 days: intraepidermal eosinophilic vesicle |
| 2 | First 24h | Neonatal period | Vesicular stage (vesicles in lower limbs, less prominent compared to sister [case 1]) | Hyperpigmented stage (no evidence of verrucous stage) | Topical corticosteroid | • Focal infantile superficial haemangioma• Supernumerary nipples | 7 months: deletion in exons 4–10 of IKBKG gene | 6 days: sample could not be assessed due to low cell count |
| 3 | 7 days | Neonatal period | Vesicular and hyperpigmented stages (vesicles in torso, lower extremities and scalp and, and hyperpigmented lesions along Blaschko's lines in lower extremities) | Hyperpigmented stage (no evidence of verrucous stage) | No | Not performed | 17 days: compatible with IP | |
| 4 | First 24h | 1 month and 15 days | Vesicular stage (vesicles along Blaschko's lines in lower limbs and top of upper limbs) | 1) Verrucous stage2) Hyperpigmented stage[Occasional exacerbation in verrucous stage] | Vertex baldness | 6 months: deletion in exons 4–10 of IKBKG gene. De novo | 1 month: eosinophilic spongiosis | |
| 5 | First 24h | Neonatal period | Vesicular stage (vesicles at the axillary and dorsal levels along Blaschko's lines) | Verrucous and hyperpigmented stages[Exacerbation with verrucous stage lesions associated with episode of acute gastroenteritis] | Fusidic acid and topical corticosteroid | • Thin and sparse hair on scalp• Thin and flat nails | 19 months: deletion in exons 4–10 of IKBKG gene | 1 month: spongiosis with high count of eosinophils in vesicle and skin |
| 6 | First 24h | Neonatal period | Vesicular stage (vesiculobullous lesions in left foot) | 1) Verrucous stage2) Hyperpigmented stage | No | Not performed | Not performed | |
| 7 | 3 days | Neonatal period | Vesicular stage (erythematous pustular lesions along Blaschko's lines in the right side of body) | Verrucous stage | No | 2 months: no changes detected by full sequencing of IKBKG gene in blood sample (PCR and MLPA) and insufficient tissue for skin testing | 10 days: spongiosis with intraepidermal blister and high eosinophil count | |
| 8 | First 24h | 6 months | Vesicular stage (vesicles distributed along Blaschko's lines in left hemithorax) | 1) Hyperpigmented stage2) Progressive resolution of lesions3) Atrophic stage | No | 11 months: deletion in exons 4–10 of IKBKG gene | Not performed | |
| 9 | First 24h | Neonatal period | Vesicular and verrucous stages (crusted blisters with a linear distribution along the upper and lower extremities) | 1) Hyperpigmented and verrucous stages2) Hyperpigmented and atrophic stages | Topical corticosteroid | Hirsutism in pubic area secondary to prolonged topical steroid therapy | 7 months: deletion in exons 4–10 of IKBKG gene. De novo | 28 days: compatible con IP |
| 10 | First 30 days | 3 months | Vesicular stage (linear vesiculobullous lesions along Blaschko's lines) | 1) Verrucous and hyperpigmented stages2) Hyperpigmented stage3) Verrucous stage.4) Atrophic stage | Topical corticosteroid | No | 3 months: deletion in exons 4–10 of IKBKG gene | 7 days: isolated necrotic keratinocytes (inconclusive result)14 days: compatible with IP |
| 11 | 15 days | 10 months | Vesicular stage (vesicles in lower extremities along Blaschko's lines) | 1) Verrucous stage2) Hyperpigmented stage3) Progressive resolution of lesions | No | 2 years: deletion in exons 4–10 of IKBKG gene | Not performed | |
| 12 | First 24h | 5 months | Vesicular stage (vesicles in linear pattern along Blaschko's lines in right lower limb, perigenital area and hemithorax) | Hyperpigmented stage | No | 5 months: variant p.Gln268Ter in IKBKG gene | Not performed | |
| 13 | First 30 days | Neonatal period | Vesicular stage (vesicles distributed along Blaschko's lines in right hemithorax, lower limbs and dorsal region) | Hyperpigmented stage | No | 2 months: variant p.Gln268Ter in IKBKG gene | Not performed |
Noncutaneous dermatologic abnormalities resulting from IP (in hair and nails) only occurred in 3 of the patients (Table 2).
Neuroimaging tests, including cranial ultrasound, computed tomography (CT) and magnetic resonance imaging (MRI) scans, were performed in all patients and revealed involvement of the brain in 6 patients, associated with clinical manifestations in only 3. In 2 of the patients, the initial cranial ultrasound did not detect any abnormalities (cases 4 and 10), but a subsequent cranial MRI scan did.
Three patients exhibited CNS involvement in the form of convulsive seizures. These same patients experienced psychomotor retardation, while the rest of the patients did not (Table 3).
Manifestations in other organs or systems in the case series.
| Case | Neuroimaging and brain function tests (age performed) | Neurologic involvement/abnormal psychomotor development | Ophthalmologic | Dental | Haematologic | Duration of follow-up (age) |
|---|---|---|---|---|---|---|
| 1 | • Cranial ultrasound (4 days and 15 days): signs of vascular calcification in a lenticulostriate artery• Cranial MRI (1 month): normal | No/normal | No abnormalities | Conical teethTooth agenesis | Eosinophilia | 3 years and 6 months |
| 2 | Cranial ultrasound: normal | No/normal | No abnormalities | Conical teeth | Eosinophilia | 3 years and 6 months |
| 3 | • Cranial ultrasound (20 days): hyperechoic lesion in left frontoparietal region compatible with gliosis or ischaemia• Cranial MRI (6 days): subacute ischaemic lesions in white substance of both cerebral hemispheres around the anterior and middle cerebral arteries, with predominance of the left side• EEG (10 days): focal spike-and-wave discharges in frontal region of right hemisphere | Neonatal period: focal status epilepticus secondary to ischaemic attack at the level of the anterior and middle cerebral arteries, bilateral with predominance of left side/left-sided hemiparesis | No abnormalities | Not assessed (lost to follow-up) | Eosinophilia | 1 month and 17 days (lost to follow-up) |
| 4 | • Cranial ultrasound (4 months): normal• Cranial MRI (3 years): subcentimetric lesions in internal capsules of both cerebral hemispheres• Cranial MRI (5 and 7 years): white matter lesions at centrum semiovale and subcortical structures of left brain | No/normal | No abnormalities | Delayed tooth eruption (1 year and 10 months)Conical teeth | Normal | 9 years and 3 months |
| 5 | • Cranial ultrasound (2 and 17 days): possible IVH in left choroid plexus with mild dilatation of left lateral ventricle• Cranial ultrasound (1½ months): resolution of IVH, no abnormalities• Cranial MRI (1½ years): normal | 3 months post birth: generalized tonic-clonic seizures/global developmental delay | Grade 1 retinopathy of prematurity that resolved by 1 year post birth | Normal | Normal | 4 years |
| 6 | Cranial ultrasound (20 days): normal | No/normal | No abnormalities | Not assessed (lost to follow-up) | Eosinophilia | 20 days (lost to follow-up) |
| 7 | Cranial ultrasound (24h and 30 days): normal | No/normal | No abnormalities | Normal | Normal | 1 year and 3 months |
| 8 | Cranial ultrasound (7 months): normal | No/normal | No abnormalities | Normal | Normal | 10 years and 8 months |
| 9 | Cranial MRI (1 year and 2 months): normal | No/normal | Haemorrhagic–ischaemic lesion in nasal peripheral retina of left eye | Normal | Normal | 12 years |
| 10 | • Cranial ultrasound (7 days): normal• Cranial MRI (15 days): hypointense winding in right parietal and occipital lobes possibly associated with venous malformations• EEG: focal paroxysmal activity in right parietotemporal region | Neonatal period: left-side seizures. Symptomatic focal epilepsy/left-sided hemiparesis | Total retinal detachment in left eyeHigh myopia in right eye | Tooth agenesis | Normal | 13 years and 6 months |
| 11 | Cranial CT: normal | No/normal | No abnormalities | Conical teeth | Normal | 14 years and 10 months |
| 12 | Cranial MRI: bilateral, nonspecific diffuse involvement of periventricular white matter in frontal and occipital regions | No/normal | Vessel tortuosity and hypopigmented lesions in both retinas | Hypodontia | Normal | 2 years and 7 months |
| 13 | • Cranial MRI: normal• EEG: normal | No/normal | Right eye: retinal bleedingLeft eye: nonspecific retinal hyperpigmentation | Conical teethTooth agenesis | Normal | 2 years and 9 months |
CT, computed tomography; EEG, electroencephalogram; IVH, intraventricular haemorrhage; MRI, magnetic resonance imaging.
The ophthalmological follow-up detected abnormalities in 5 cases, all involving the retina, and associated with preterm birth in 1 case (Table 3).
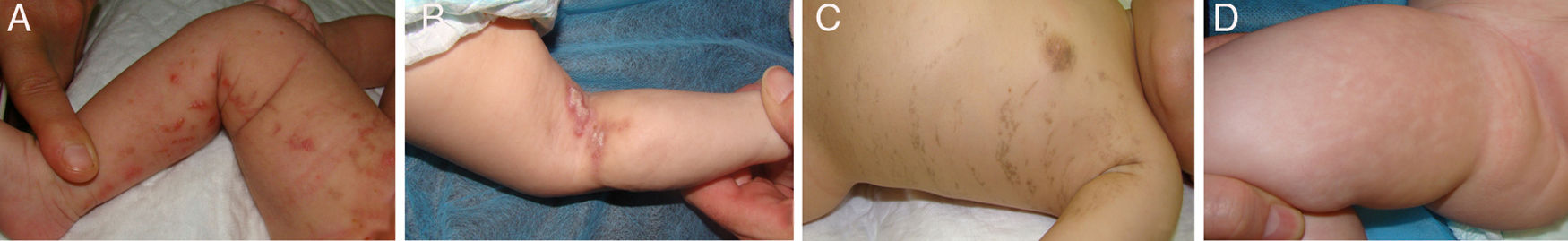
Dental abnormalities occurred in 7 of the 11 patients that remained in follow-up (in cases 3 and 6, patients were lost to follow-up before tooth eruption), and included abnormalities in the number or morphology of the teeth and delays in tooth eruption (Table 3 and Fig. 2).
Laboratory tests detected significant abnormalities in blood (eosinophilia), with no other relevant findings in 4 of the patients (cases 1, 2, 3 and 6) (Table 3).
Eleven patients underwent molecular genetic testing of peripheral blood samples, which detected deletions in exons 4–10 of the IKBKG gene in 8 cases (targeted PCR amplification to identify deletions in exons 4–10), no changes in this gene in 1 patient (case 7) and the pathogenic variant p.Gln268Ter (c.802C>T) of the IKBKG gene in the heterozygous state in 2 patients (cases 12 and 13).
The first molecular test performed in case 13 (analysis of deletions in exons 4–10 by means of long-range PCR) did not find any changes. For this reason, the IKBKG gene was sequenced (Sanger sequencing of coding regions and spicing sites of the IKBKG gene for subsequent comparison with the reference sequence NM_003639.3), which led to detection of the pathogenic variant p.Gln268Ter (c.802C>T), which produces a premature stop codon and therefore a truncated protein that may not be functional and causing the disease.
Cases 12 and 13 corresponded to patients born after in vitro fertilization of a donor egg in the same fertility clinic. This fact arose suspicion that both eggs could have come from the same donor, for which reason genetic testing in case 12 was aimed from the beginning to ruling out the pathogenic variant p.Gln268Ter (c.802C>T).
In case 7, due to a clinical presentation compatible with mosaicism (lesions restricted to a single portion of the body) and a negative result of molecular testing of blood samples (IKBKG sequencing after amplification PCR of coding exons and splicing sites and comparison with reference sequence NM_003639.3, and deletion and insertion analysis by means of Multiplex Ligation-dependent Probe Amplification [MLPA] assay), genetic testing of a skin biopsy sample was attempted, but could not be performed due to technical difficulties.
The mean age at the time of molecular genetic testing was 8 months. A skin biopsy sample was collected in 8 patients, always in the first 30 days from birth, with features compatible with IP (eosinophilic spongiosis) in all cases (Table 2).
The mothers of 9 patients underwent genetic testing, with detection of the same mutation found in the index case in 7. The other 2 mothers had a clinical diagnosis of IP and reported that the results of genetic testing (performed in a different laboratory outside Spain) were compatible with the disease, but did not produce the corresponding reports.
DiscussionOur study includes a series of cases of IP for which we collected all the available data on the clinical course of the patients, including photographic, genetic and laboratory evidence. It evinced the considerable variation in the documented clinical manifestations associated with IP in different organs and systems.
Mutations in the IKBKG gene consisting of deletions in exons 4–10 were found in most of the cases under study, in agreement with the previous literature (80–90%).1,2 In our case series, sequencing of the IKBKG gene led to the identification of a novel mutation not described to date (pathogenic variant p.Gln268Ter [c.802C>T]). This underscores the importance of sequencing this gene in every case in which targeted analysis of deletions in exons 4–10 turns out to be negative.
Within our case series, we ought to highlight the presence of 1 case of somatic mosaicism (case 7). In these cases, if genetic testing of blood samples with sequencing of the entire IKBKG gene (including deletion and insertion analysis using MLPA) gives negative results, performance of genetic testing in samples of skin affected by the disease is recommended.12
Since this disease is lethal in males, it should be suspected in mothers with recurrent miscarriage of male foetuses, a history that was present in the mothers of some of the patients in our series.6,8,13
Due to the X-linked dominant pattern of inheritance of IP, genetic testing of the mother is recommended if the mother has not been evaluated in the past. However, it is important to take into account that 65% of IP cases result from de novo mutations.2 The percentage in our series was lower, as de novo mutations only occurred in 2 of the patients under study (18%, cases 4 and 9).
In agreement with previous studies, blistering (vesicular stage) was the first cutaneous manifestation to develop in all cases at onset in the neonatal period, in some cases overlapping with other stages.2,12
Outbreaks of skin lesions may develop without an apparent cause, but in some instances may be associated to intercurrent infection, as occurred in one of our patients.
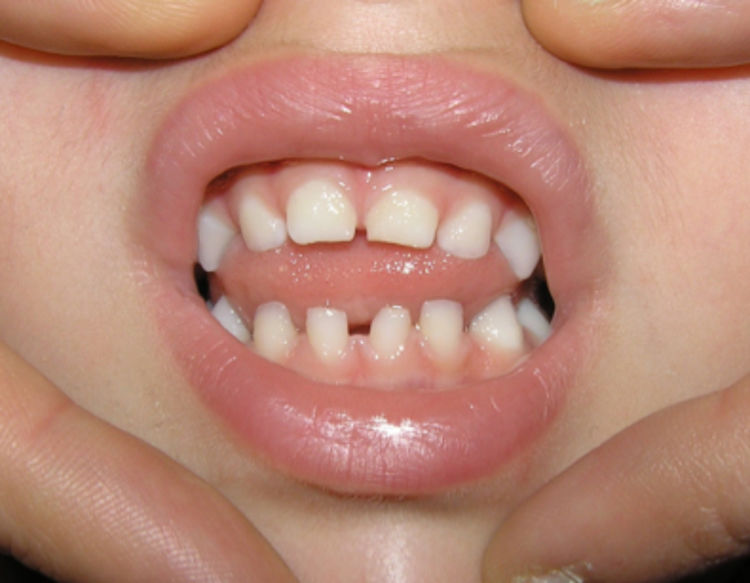
Only 2 patients in the case series had hair anomalies (15.3%, cases 4 and 5) in the form of cicatricial alopecia and thinning hair on the scalp, a proportion that was lower compared to previous studies (>50%)1–3,14 (Fig. 3).
Nail lesions may affect between 7% and 40% of patients with IP and develop between ages 3 and 45 years (mean, 26 years).1,3,7 In our study they only occurred in 1 patient (7%, case 5); however, these are abnormalities that develop with age, so monitoring for their future development must continue. In some cases, at later ages, patients may develop painful keratotic subungual or periungual tumours that cause pain or deformities caused by osteolysis of the underlying phalanxes, and, in very rare cases, subungual squamous cell carcinoma.1,3,7,12
Central nervous system (CNS) manifestations occur in 30–50% of patients with IP, with development of heterogeneous lesions in the brain (infarction or involvement of white or grey matter at different levels). Although their pathogenesis has not been fully elucidated, it appears to be related to vasculopathy in the brain resulting from the inflammatory activity associated with the disease.1,2,9,15 Our findings were consistent with the previous literature, as some form of CNS abnormality was detected in 50% of patients evaluated by means of diffusion-weighted MRI of the head.
Cranial ultrasonography can be used for initial assessment in the neonatal period, although MRI can contribute more information, especially using the diffusion-weighted imaging mode.2,15
In some cases, the initial cranial ultrasound may be normal, but subsequent cranial MRI scans may reveal the presence of lesions, as was the case of 2 patients in our study (cases 4 and 10). This may suggest that the lesions developed sometime between performance of the ultrasound scan and performance of the MRI scan, false negatives of ultrasound examination due to technique limitations or incorrect interpretation, or to the higher sensitivity of MRI compared to ultrasonography. On the other hand, we ought to highlight that ultrasound detected signs of vascular calcification in case 1, whereas the subsequent cranial MRI scan was normal. These findings suggest that transfontanellar ultrasound may not suffice for neurologic assessment of patients with IP, and that a cranial MRI scan should be performed as early as possible. Some studies recommend performance of magnetic resonance angiography for a more accurate assessment of the degree of vasculopathy.15,16
Neurologic changes manifest in the form of seizures, intellectual disability, motor impairment (paresis/spastic paralysis) or microcephaly.1,2,9,10,15 In the neonatal period, the clinical presentation of CNS involvement usually consists of seizures or altered level of consciousness.15
In our series, only 3 patients experienced seizures (cases 3, 5 and 10), and they were the same patients that developed other neurologic abnormalities (hemiparesis in 2 and psychomotor development in 1).
We only found an association between imaging tests and neurologic manifestations in 2 cases (3 and 10); the rest of the girls with abnormal findings in imaging tests were clinically asymptomatic. However, these patients are still young, so it is important that we continue to monitor their neurodevelopment to further clarify this finding.
Eye anomalies occur in approximately 20–35% of patients with IP (wide range depending on the study: 16–77%),1,7 and are frequently associated with neurologic changes, with blindness in 7% of patients. It is believed that the mechanisms underlying the damage to the retina involve tissue ischaemia resulting from vaso-occlusive events.1,2,17
Our study identified some form of eye anomaly associated with IP in 4 cases (30%), and neurologic abnormalities in only 1 patient (case 10). Most of the patients with retinal involvement required laser treatment, and 1 required surgery for a unilateral detached retina and eventually enucleation of the eye. It is particularly important to be careful in the diagnosis of retinopathy of prematurity, as occurred in case 5, as it may be confused with anomalous development of retinal vessels.14 Our patients did not exhibit nonretinal eye anomalies associated with IP (strabismus, optic nerve atrophy, ptosis, iris hypoplasia, conjunctival pigmented lesions, microphthalmia, vortex keratitis, cataracts, nystagmus and uveitis).1,14
Dental abnormalities occur in 50–80% of affected individuals, most frequently peg teeth, followed by hypodontia, tooth agenesis, delayed tooth emergence and loss of teeth.1,11 Our findings were consistent with this (abnormalities found in 63% of patients that underwent a dental evaluation). There are other abnormalities of the oral cavity associated with IP, most frequently cleft palate and high-arched palate, although dental malocclusion and facial asymmetry can also occur (Fig. 2).1,2,11
Breast anomalies have been described in 11–30% of patients with IP, most frequently supernumerary nipples, but also possibly hypoplasia or aplasia.3,7,12 In our series, breast anomalies only happened in 1 patient that had a supernumerary nipple (case 2).
Another finding associated with IP is leucocytosis with eosinophilia, especially during stages 1 and 2.7,12,17 Our retrospective review of cases revealed eosinophilia in 4, in every case in the neonatal period and in most cases during stages 1 or 2. This test was performed when patients required it for conditions other than IP (risk of infection in neonatal period, infectious disease, etc.).
Different studies have described patients with IP with autoimmune disorders, immunodeficiencies and malignancies, which is related to the modulator role of NF-kB.3,18,19
The patients in our series did not exhibit any such comorbidities; however, it is necessary to continue the follow-up to watch for their potential development.
Our study was limited by the loss to follow-up of several patients who moved to other cities or did not return to the clinic for follow-up; another limitation is the young age of the patients, which did not allow us to obtain information on abnormalities that develop through the years.
We found that diagnosis of IP occurred earlier in the cases detected in the past 5 years, probably due to an increased knowledge of the disease. It is important to establish a comprehensive follow-up plan covering the lifespan of the patient, including preconception and prenatal genetic counselling.16 The purpose of such a plan is to detect the signs and disorders associated with IP to prescribe targeted treatment as soon as possible and prevent complication, especially those involving the CNS and eyes, which are determinants of the prognosis of IP.2,10,20
In conclusion, we present the most detailed study published to date on the subject of IP in Spain, with findings similar to those of larger case series published in the international literature, which highlights the importance of providers being aware of this disease and of carrying out a multidisciplinary evaluation and follow-up.
Conflicts of interestThe authors have no conflicts of interest to declare.
We thank the Department of Genetics of the Hospital Materno Infantil, Malaga, Spain.
Please cite this article as: Ocaña Jaramillo S, del Boz J, Vera Casaño Á. Incontinencia pigmenti. Estudio descriptivo de la experiencia en dos centros hospitalarios. An Pediatr (Barc). 2020;92:3–12.
