





Ketogenic dietary therapies (KDT) produce anticonvulsant and neuroprotective effects, reduce seizures and improve the cognitive state in patients with epilepsy. Our purpose was to evaluate the effects of KDT in children with refractory epilepsy (effectiveness, side effects, impact on nutritional status and growth).
MethodsA retrospective and prospective observational descriptive study was conducted in a Spanish tertiary hospital (January 2000 to December 2018). One hundred sixty pediatric patients with epilepsy were treated with KDT (82 males; mean age 5 years 9 months). Seizures, anti-epileptic drugs, anthropometric measures, side effects, and laboratory assessment were monitored baseline and at 3, 6, 12 and 24 months after the onset of KDT.
ResultsIn these time intervals, the seizure-free patients were: 13.7, 12.5, 14.4 and 10.6%, respectively, and a reduction of seizures ≥ 50% was achieved in 41.9, 37.5, 28.7 and 16.2%. Side effects were frequent, especially digestive disorders, hypercalciuria, hypoglycemia, hepatic dysfunction and dyslipidemia. Prealbumin, retinol binding protein, vitamin A and magnesium decreased significantly. Height was affected, especially in children below 2 years.
ConclusionsKDT are effective for refractory epilepsy in children. However, adverse effects are frequent, and it may affect nutritional status and growth.
Las terapias dietéticas cetogénicas (TDC) tienen efecto neuroprotector y anticonvulsivante, reducen las crisis epilépticas y mejoran el estado cognitivo en pacientes epilépticos. Nuestro propósito fue evaluar los efectos de las TDC en niños con epilepsia refractaria (eficacia, efectos secundarios, impacto en el estado nutricional y crecimiento).
MétodosSe realizó un estudio observacional descriptivo retrospectivo y prospectivo en un hospital terciario español (enero de 2000-diciembre de 2018). Ciento sesenta pacientes pediátricos con epilepsia fueron tratados con TDC (82 varones; edad media 5 años 9 meses). Las convulsiones, los fármacos antiepilépticos, la antropometría, los efectos secundarios y los parámetros analíticos se controlaron al inicio del tratamiento y a los 3, 6, 12 y 24 meses.
ResultadosEn estos intervalos los pacientes libres de crisis fueron: 13,7%, 12,5%, 14,4% y 10,6%, respectivamente, lográndose una reducción de las convulsiones ≥ 50% en el 41,9%, 37,5%, 28,7% y 16,2%. Los efectos secundarios fueron frecuentes, especialmente trastornos digestivos, hipercalciuria, hipoglucemia, disfunción hepática y dislipidemia. La prealbúmina, la proteína de unión al retinol, la vitamina A y el magnesio disminuyeron significativamente. La talla se vio afectada, especialmente en niños menores de 2 años.
ConclusionesLas TDC son efectivas para la epilepsia refractaria infantil. Sin embargo, los efectos adversos son frecuentes y pueden afectar al estado nutricional y al crecimiento.
Epilepsy is one of the most prevalent neurologic disorders of childhood. Antiseizure medicines (ASM) improve control of seizures in most patients. However, up to one third of them (7%-20% of children and 30%-40% of adults)1,2 may have refractory epilepsy, possibly associated with cognitive impairment, adverse events (AEs) and poor quality of life. In these cases, ketogenic dietary therapies (KDT) may be an alternative option.
There are different types of KDT.3 The classic ketogenic diet (CKD) provides 87% to 90% of the total energy intake in the form of lipids.3 The intake of carbohydrate and protein varies from ratios of 3:1 to 4:1 (3 to 4 grams of lipids per gram of carbohydrate and protein). In the medium-chain triglyceride diet (MCT, MCT-KD), lipids contribute 71% of the total energy intake (11% in the form of natural lipids and 60% in the form of MCTs).4 The modified MCT-KD allows a higher proportion of natural lipids (41%). The modified Atkins diet (MAD) allows unrestricted consumption of protein and fat and restricts the intake of carbohydrate (10 g/day).5 The low-glycaemic index diet (LGID) allows a more liberal total carbohydrate intake (40-60 g/day) but restricted to foods with a glycaemic index of less than 50.6 There is no clear evidence indicating differences in efficacy between the CKD, MCT-KD or MAD.8 Some studies suggest that the LGID is also efficacious,9 despite not inducing ketosis, and a recent randomised clinical trial found that it was not inferior to the CKD.10
Ketogenic diets can cause gastrointestinal,11,12 hepatic12 and renal problems,13,14 dyslipidaemia,15 nutrient deficiencies16,17 and changes in growth.18–22
The aim of our study was to assess the effectiveness of KDT, associated adverse events and their impact on growth and nutritional status. We compared different KDT and age groups to identify differences in the variables under study.
Sample and methodsWe collected anonymised data for every patient with epilepsy aged less than 18 years treated with a KDT in a tertiary care hospital between January 2000 and December 2018. The study had a retrospective and prospective observational descriptive design (starting in May 2015) and was approved by the Clinical Research Ethics Committee of the Hospital Infantil Universitario Niño Jesús (R-0002/15). Patients (or their families) were informed about the study, signed an informed consent form and underwent assessments at baseline and at 3, 6, 12 and 24 months.
The calibration of the diets was based on the energy intake recommendations of the World Health Organization (WHO)23 for age and weight if indirect calorimetry was not performed to assess energy expenditure. The protein intake was calculated based on current recommendations (1 g/kg/day in children > 1 year and 1.5 in infants).24 The Holliday-Segar method was used to estimate the maintenance water requirements,25 although at least the minimum recommended by the WHO was administered in every case.26 Trace minerals, vitamins and minerals in formulations devoid of carbohydrates were prescribed to ensure the recommended daily intake26,27 if there was evidence of low intake or nutrient deficiency.
We assessed efficacy based on the reduction in the use of ASM and the percent reduction in seizures relative to baseline: 100% (seizure-free), 90-100%, 50-90%, < 50%, 0% (no improvement) or worsening.
We classified AEs as early (first month) or late. We analysed the weight, height and body mass index adjusted for sex and age based on the WHO growth standards. We considered the height normal if the height z-score was between –2 and +2, mildly low if the z-score was less than –2 and equal or greater than −3 and very low if it was less than −3. The laboratory workup included a complete blood count, blood chemistry and lipid panels, blood gas analysis, urine sediment examination and urinary calcium, protein and citrate/creatinine ratios, and levels of prealbumin, retinol-binding protein, vitamins A, D, E, B9, B12, zinc, selenium, parathyroid hormone and carnitine.
The statistical analysis was performed with the software SPSS Statistics version 16.0. We summarised quantitative data as mean and standard deviation (SD) if normally distributed and otherwise as median and range or interquartile range. Qualitative data were summarised as frequency distributions. The analysis was by intention to treat.
We compared the efficacy of 3 diet groups: CKD (3:1 and 4:1 ratios), MCT-KD (classic or modified) and MAD. To detect differences in anthropometric parameters at baseline and at different time intervals during the follow-up based on the type of KDT and age group (< 2 years, 2-5 years, 5-10 years, > 10 years) we performed a repeated-measures analysis of variance (ANOVA) for paired data. We used the Fisher exact test to compare the efficacy of different KDT. We used the Student t test for paired samples to compare laboratory values at baseline and at 12 and 24 months. We used ANOVA and the Student t test for independent samples to compare these indicators by type of KDT. Statistical significance was defined as a p-value of 0.05 or less.
ResultsKDT were used for treatment of 160 patients, including 71 from May 2015. Table 1 presents the characteristics of the patients. All had refractory epilepsy except 1 patient that had not received ASM due to suspicion of glucose transporter type 1 (GLUT1) deficiency syndrome. Table 2 presents the causes of epilepsy. The median age at onset of epilepsy was 291 days (interquartile range, 936.2). Ninety percent of the patients had seizures daily. Before initiation of KDT, patients were receiving a median of 6 antiepileptic drugs (range, 0-16), and 59% experienced AEs. Other treatments used in the patients were vagus nerve stimulation (5), surgery (7), steroid therapy (48), immunoglobulin (15) and other, such like supplementation with vitamin B6 (47%). Twelve had previously been managed with a KDT, of who 5 had responded well.
Characteristics of the patients in the study and of the ketogenic diet interventions.
| Patients with KD interventions (n = 160) | |
|---|---|
| Sex | Male, 82 (51%) |
| Female, 78 (49%) | |
| Psychomotor retardation/intellectual disability | 73% |
| Type of seizures | Generalised tonic-clonic, 10 (6.2%) |
| Myoclonic, 27 (16.9%) | |
| Tonic, 30 (18.7%) | |
| Focal, 37 (23%) | |
| Atonic, 3 (1.9%) | |
| Absence, 7 (4.4%) | |
| Clonic, 6 (3.7%) | |
| Various, 40 (25%) | |
| Age at onset of epilepsy | Newborn, 21 (13.1%) |
| Infant, 68 (42.5%) | |
| 1-5 years, 54 (33.7%) | |
| 5-10 years, 12 (7.5%) | |
| > 10 years, 5 (3.1%) | |
| Number of seizures before initiation of KDT | ≤ 1/day, 16 (10%) |
| 2-10/day, 89 (55.6%) | |
| > 10/day, 35 (21.9%) | |
| Countless, 3 (1.9%) | |
| Unknown, 5 (3.1%) | |
| Status epilepticus, 12 (7.5%) | |
| Age at initiation of CKD | Infant, 25 (15.6%) |
| 1-2 years, 15 (9.4%) | |
| 2-5 years, 45 (28.1%) | |
| 5-10 years, 40 (25%) | |
| > 10 years, 35 (21.9%) | |
| Type of CKD | CKD, 3:1; 68 (42.5%) |
| CKD, 4:1; 16 (10%) | |
| DMAD, 58 (36.2%) | |
| MCT-KD, 2 (1.2%) | |
| Modified MCT-KD, 16 (10%) | |
| Route of administration | Oral, 120 (75%) |
| NGT, 24 (15%) | |
| Gastrostomy, 16 (10%) | |
| Median duration of CKD (range) | 259 days (5 days-9 years and 6 months) |
| Reason for discontinuation of CKDa | Ineffective, 76 (60.3%) |
| Non-adherence, 17 (13.5%) | |
| Adverse events, 11 (8.7%) | |
| Duration > 2 years, 10 (8%) | |
| Lost to follow-up, 5 (4%) | |
| Death, 3 (2.5%) | |
| Other, 4 (3%) |
CKD, classic ketogenic diet; MCT, medium-chain triglycerides; NGT, nasogastric tube.
Aetiology of epilepsy.
| Aetiology | n (%) | Specific cause |
|---|---|---|
| Unknown | 29 (18.1%) | |
| Metabolic | 24 (15%) | 11: GLUT1-DS (5 with genetic confirmation) |
| 9: mitochondrial disease | ||
| 2: neuronal ceroid lipofuscinosis (identified a posteriori) | ||
| 1: severe neonatal hypoglycaemia | ||
| 1: PDH deficiency | ||
| Structural | 43 (26.9%) | 22: malformations of cortical development (dysplasia in 15; lissencephaly in 3; polymicrogyria in 2; hemimegaloencephaly in 1; subcortical heterotopia in 1) |
| 16: hypoxic-ischaemic brain injury (protein C deficiency in 1; factor VIII variant in 1; meningitis in 1; sepsis in 1; leukaemia in 1; perinatal disease in 9; ischaemic stroke in 1; complication of complex cardiac surgery in 1) | ||
| 2: cortical tubers (tuberous sclerosis complex) | ||
| Complex cerebral malformation (macrencephaly and neuronal migration disorder [polymicrogyria and periventricular nodular heterotopias]) and giant arachnoid cyst | ||
| Agenesis of corpus callosum | ||
| 1: leptomeningeal angioma (Sturge-Weber syndrome) | ||
| Genetic | 39 (24.4) | 28: gene variants (involving CDKL5 in 6; SCN1A in 4 [Dravet syndrome]; KCNQ2 in 3; STXBP1 in 3; SLC9 in 2; X-linked MECP2 in 2; SCN2A in 1 (early infantile epileptic encephalopathy); CHRNA4 in 1 (nocturnal temporal lobe epilepsy); SCN1B in 1; RBFOX1 in 1; CACNA1H in 1; CNTNAP2 in 1 (associate d with dysplasia and Pitt-Hopkins-like syndrome); PTPN11 in 1 (Noonan syndrome); PHOX2B in 1(congenital central hypoventilation syndrome) |
| 11: chromosomal disorders (trisomy 21/Down syndrome in 3; inverted duplication of proximal chromosome 15 in 3; 1 p deletion in 1; 11q14 deletion in 1; Angelman syndrome in 1; ring chromosome 17 in 1; 16p duplication in 1) | ||
| Infectious | 6 (3.7) | 3: herpes simplex virus |
| 2: Streptococcus pneumoniae | ||
| 1: cytomegalovirus (perinatal) | ||
| Immune | 4 (2.5) | 2: confirmed (positive anti-NMDA antibodies) |
| Genetic syndrome | 15 (9.4) | 3: Lennox-Gastaut syndrome |
| 3: West syndrome | ||
| 3: FIRES | ||
| 4: Doose syndrome | ||
| 1: Ohtahara syndrome | ||
| 1: Landau-Kleffner syndrome |
CACNA1H, calcium voltage-gated channel subunit alpha 1 H; CDKL5, cyclin dependent kinase-like 5; CHRNA4, cholinergic receptor nicotinic alpha 4 subunit; CNTNAP2, contactin-associated protein-like 2; FIRES, febrile infection-related epilepsy syndrome; KCNQ2, potassium voltage-gated channel subfamily Q member 2; MECP2, methyl-CpG binding protein 2; NMDA, N-methyl-D-aspartate; PDH, pyruvate dehydrogenase; PHOX2B, paired-like homeobox 2B; RBFOX1, RNA binding fox-1 homolog 1; SCN1A, sodium voltage-gated channel alpha subunit 1; SCN2A, sodium voltage-gated channel alpha subunit 2; SCN1B, sodium voltage-gated channel beta subunit 1; GLUT1-DS, Glucose transporter type 1 deficiency syndrome; SLC9, solute carrier family 9; STXBP1, syntaxin binding protein 1; PTPN11, protein tyrosine phosphatase non-receptor type 11.
The mean age at initiation of the KDT was 5 years and 9 months (range, 79 días-17 years and 4 months). The mean time elapsed from onset to initiation of the KDT was 3 years and 10 months (range, 7 days-14 years and 6 months).
Before initiation of the KDT, some abnormalities in markers of nutrition were detected (Table 3), and the intake of vitamins and trace minerals was deficient in 16.8% of patients.
Trends in serum and urine markers.
| Reference range | Mean (SD) | Number of patients above or below the reference range (%)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Timepoint (n) | Baseline (160) | 3 m (126) | 6 m (95) | 12 m (62) | 24 m (32) | Baseline | 3 m | 6 m | 12 m | 24 m | |
| Serum nutritional markers | |||||||||||
| Prealbumin (mg/dL) | 15-40 | 21.3 (6.5) | 18.5 (4.7) | 18.4 (4.5) | 19 (6.2) | 17.8 (3.8) | 16 (10) | 21 (16.6) | 19 (20) | 15 (24.2) | 7 (21.9) |
| RBP (mg/dL) | 2.5-6.9 | 3.5 (1.2) | 2.6 (1.3) | 2.6 (1.1) | 2.7 (1.0) | 2.6 (0.7) | 32 (20) | 20 (15.9) | 22 (23.2) | 20 (32.3) | 7 (21.9) |
| Ferritin (ng/mL) | 7-140 (< 15 y) | 46.8 (57) | 42.6 (43) | 51.9 (44) | 58.4 (37) | 55.3 (39.7) | 4 (2.5) | 3 (2.4) | 0 | 0 | 1 (3) |
| 15-175 (≥ 15 y) | |||||||||||
| Micronutrients | |||||||||||
| Magnesium (mg/dL) | 1.5-2.5 | 2.1 (0.3) | 2.0 (0.2) | 2.1 (0.2) | 2.0 (0.2) | 1.97 (0.1) | 1 (0.6) | 1 (0.8) | 0 | 0 | 0 |
| Selenium (μg/L) | 70-120 | 71.2 (21) | - | 87.7 (14) | 85.7 (21) | 87.5 (14.5) | 31 (19.3) | - | 2 (2.1) | 4 (6.4) | 1 (3) |
| Zinc (μg/dL) | 70-150 | 91.2 (32) | - | 102.5 (24) | 95.7 (24) | 101.4 (17.3) | 12 (7.5) | - | 2 (2.1) | 4 (6.4) | 0 |
| Total carnitine or levocarnitine (μmol/L) | 21.5-64.5 | 55.6 (25) | - | 84.8 (47) | 81.2 (40) | 69 (31.4) | 17 (10.6) | - | 2 (2.1) | 5 (8) | 1 (3) |
| 20-50 | 42.1(18) | - | 47.2 (24) | 46.5 (18) | 47 (16.7) | ||||||
| Vitamin A (mg/L) | 0.2-0.6 | 0.43 (0.2) | 0.37 (0.1) | 0.35 (0.1) | 0.35 (0.1) | 0.36 (0.2) | 3 (1.9) | 1 (0.8) | 1 (1) | 4 (6.4) | 1 (3) |
| Vitamin E (mg/L) | 3-9 (< 12 y) | 5.6 (3.9) | 4.6 (1.8) | 5.1 (1.2) | 4.8 (1.4) | 5.0 (1.2) | 9 (5.6) | 3 (2.4) | 4 (4.2) | 6 (9.7) | 2 (6.2) |
| 5-20 (≥ 12 y) | |||||||||||
| Folic acid (B9) (ng/mL) | 3.9-23.9 | 12.1 (6.2) | 16.6 (11) | 16.9 (5.8) | 19.1 (6.4) | 17 (5.4) | 5 (3.1) | 0 | 0 | 0 | 0 |
| Vitamin B12 (pg/mL) | 250-914 | 814 (624) | 955 (559) | 876 (355) | 897 (533) | 916 (489) | 3 (1.9) | 0 | 0 | 0 | 0 |
| 25-hydroxivitamin D (ng/mL) | 20-80 | 28.6 (11) | 35.8 (12) | 37.8 (14) | 34.4 (14) | 31 (10) | 25 (15.6) | 2 (1.6) | 3 (3.2) | 5 (8) | 3 (9.4) |
| Calcium (mg/dL) | 8.8-10.8 | 9.6 (0.4) | 9.6 (0.4) | 9.6 (0.4) | 9.6 (0.4) | 9.6 (0.3) | 3 (1.9) | 0 | 1 (1) | 0 | 0 |
| Phosphorus (mg/dL) | 4.5-6.5 | 5.0 (0.8) | 4.9 (0.7) | 4.9 (0.7) | 4.7 (0.7) | 4.8 (0.5) | 31 (19.4) | 24 (19.0) | 20 (21) | 24 (38.7) | 9 (28) |
| Lipid profile | |||||||||||
| Cholesterol (mg/dL) | 100-200 | 164 (37) | 195 (61) | 197 (53) | 196 (53) | 195 (44) | 23 (14.4) | 39 (31) | 32 (33.7) | 27 (43.5) | 14 (43.7) |
| Triglycerides (mg/dL) | 35-115 | 81 (50) | 117 (87) | 108 (61) | 115 (95) | 88 (60) | 27 (16.9) | 27 (21.4) | 20 (21) | 10 (16.1) | 3 (9.4) |
| Liver function markers | |||||||||||
| AST (U/L) | 28-50 | 33.2 (16) | 30.1 (14) | 31.7 (17) | 33.7 (35) | 27.7 (6.7) | 16 (10) | 7 (5.5) | 6 (6.3) | 5 (8) | 0 |
| ALT (U/L) | 15-50 | 19.3 (16) | 20.6 (14) | 20.5 (13) | 23.7 (38) | 19 (8.2) | 4 (2.5) | 3 (2.4) | 4 (4.2) | 2 (3.2) | 0 |
| GGT (U/L) | 8-30 | 52.9 (110) | 33.7 (65) | 31.5 (56) | 27.9 (23) | 27 (24.4) | 56 (35) | 18 (14.3) | 12 (12.6) | 13 (21) | 6 (18.7) |
| Blood gas analysis | |||||||||||
| pH | 7.35-7.45 | 7.4 (0.04) | 7.3 (0.03) | 7.3 (0.04) | 7.3 (0.04) | 7.3 (0.04) | 6 (3.7) | 7 (5.5) | 5 (5.3) | 5 (8) | 3 (9.4) |
| Bicarbonate (mmol/L) | 21-29 | 24.4 (6.5) | 22.3 (3.5) | 27.7 (7.9) | 21.9 (3.8) | 21.5 (2.9) | 12 (7.5) | 24 (19.0) | 8 (8.4) | 13 (21) | 7 (21.9) |
| Hormones | |||||||||||
| IGF-1 (ng/mL) | 49-288 | 108 (77) | - | 117 (110) | - | 81.7 (49.5) | 10 (6.2) | - | 9 (9.5) | - | 4 (12.5) |
| PTH (pg/mL) | 15-80 | 38.9 (22) | 27.0 (11) | 32.4 (14) | - | 30.6 (12.2) | 6 (3.7) | 2 (1.6) | 4 (4.2) | 3 (4.8) | 2 (6.2) |
| Urinary markers | |||||||||||
| Calcium/creatinine (mg/mg) | ≤ 0.8 (0-6 m) | 0.16 (0.17) | 0.3 (0.25) | 0.26 (0.24) | 0.27 (0.33) | 0.29 (0.25) | 19 (11.9) | 40 (31.7) | 27 (28.4) | 16 (25.8) | 15 (46.9) |
| ≤ 0.6 (6-12 m) | |||||||||||
| ≤ 0.2 (≥ 1 y) | |||||||||||
| Protein/creatinine (mg/mg) | ≤ 0.5 (< 2 y) | 0.24 (0.15) | 0.33 (0.19) | 0.24 (0.16) | 0.31 (0.44) | 0.23 (0.28) | 12 (7.5) | 6 (4.8) | 5 (5.3) | 4 (6.4) | 6 (18.7) |
| ≤ 0.2 (≥ 2 y) | |||||||||||
| Citrate/creatinine (mg/mg) | ≥ 0.4 | 0.8 (0.63) | 1.3 (2.7) | 0.8 (0.7) | 0.9 (0.9) | 0.6 (0.35) | 12 (7.5) | 9 (7.1) | 7 (7.4) | 11 (17.7) | 4 (12.5) |
| Ketone bodies | |||||||||||
| Beta-hydroxybutyrate (mmol/L) | > 2.4 | - | 3.1 (1.8) | 3.0 (1.7) | 3.5 (1.7) | 3.0 (1.7) | - | 47 (37.3) | 29 (30.5) | 27 (43.5) | 15 (46.9) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transpeptidase; IGF-1, insulin-like growth factor 1; m, months; PTH, parathyroid hormone; RBP, retinol-binding protein; y, years.
Table 1 presents the diets used. Between 2009 and 2014, dietary management was based on the recommendations of the International Ketogenic Diet Study Group.28 From 2015, it was based on the in-house protocol of the department of nutrition of the hospital. The CKD was used in patients fed through a nasogastric tube (NGT) or a gastrostomy tube, aged less than 2 years or with status epilepticus. The MAD was used in children older than 2 years. The diet was introduced gradually, increasing the ketogenic ratio progressively and adapting it to patient tolerance and AEs, with no restrictions to water intake.
All patients under 2 years (save 2) started the CKD with a 3:1 ratio. The MAD and MCT-KD were only prescribed to patients older than 2 years. The LGID was prescribed to 4 patients to improve adherence. The MAD was implemented via the oral route in every patient. All patients that required tube feeding received the CKD (except for 1 managed with the modified MCT-KD). Twenty-five percent of patients started the dietary intervention at the outpatient level (all with the MAD). Fasting was used in 15 patients (12 with the MCT-KD that started the diet in 2006 or earlier, and 3 treated with the CKD in the context of severe seizures). Calory restriction was implemented in 4 children that started the dietary intervention before 2014 (75%-83% of the total recommended daily intake). The mean time elapsed to achievement of ketosis (defined as a concentration of > 2.4 μmol/L) was 4.3 days (SD, 3.9; range, 12 hours-30 days). The levels of beta-hydroxybutyrate were significantly higher in patients receiving the CKD throughout the follow-up, except at the 6-month timepoint.
Efficacy of therapeutic ketogenic dietsTable 4 presents efficacy data for the sample overall, and Fig. 1 by age group. Four infants did not complete 3 months (2 due to ineffectiveness and 2 to death of the patient).
Efficacy of ketogenic dietary interventions.
| Efficacya | 3 months (n = 126) | 6 months (n = 95) | 12 months (n = 62) | 24 months (n = 32) |
|---|---|---|---|---|
| Total patients who responded | 67 (41.9%) | 60 (37.5%) | 46 (28.7%) | 26 (16.2%) |
| Favourable response | 31 (19.4%) | 24 (15%) | 13 (8%) | 7 (4.4%) |
| Excellent response | 14 (8.7%) | 16 (10%) | 10 (6.2%) | 2 (1.2%) |
| Seizure-free | 22 (13.7%) | 20 (12.5%) | 23 (14.4%) | 17 (10.6%) |
We estimated the efficacy of the diet by comparing the frequency of seizures in each interval of the follow-up to the frequency before starting the diet. We defined response to the diet as a reduction of at least 50% in the number of seizures. We defined favourable response as a reduction of 50% to 90%, an excellent response as a reduction greater than 90% and seizure-free as no epileptic seizures. The percentages were calculated by intention to treat in relation to the entire sample (160 patients).
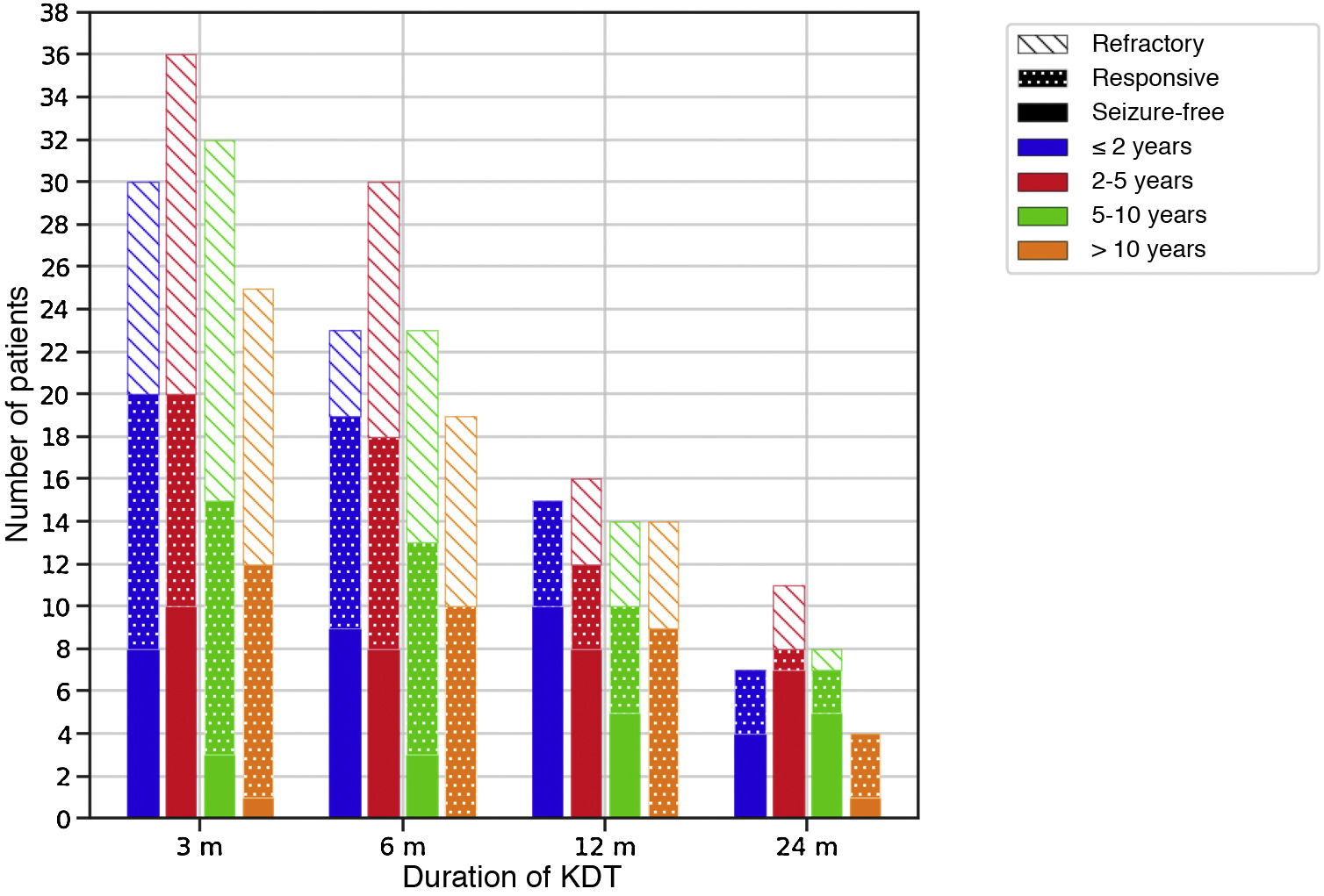
Efficacy of therapeutic ketogenic diets throughout the follow-up based on age at initiation of the diet.
The colour of the bars refers to the age of the patients at initiation of the diet and the filling to the response. Response to treatment was defined as a reduction in seizures of at least 50% compared to baseline.
m, months.
The CKD was most effective at 3 and 12 months (P < .05). Based on the aetiology of the seizures, KDT were effective in 45-10% of cases of structural aetiology, 79-47% of cases of metabolic aetiology and 59-19% of cases of genetic disorders throughout the follow-up. The use of ASM decreased in each of the time intervals in 30, 13, 9 and 9 children, respectively. The percentage of patients that took 2 or fewer AEM increased from 34% at baseline to 41-56% throughout the follow-up.
The KDT was discontinued in 126 (Table 1 and Fig. 2). Ineffectiveness was the most frequent reason for discontinuation in every age group and type of diet, followed by non-adherence in children managed with the MAD (17/12) and AEs in children managed with CKD (6/11). No child under 2 years discontinued the diet due to non-adherence.
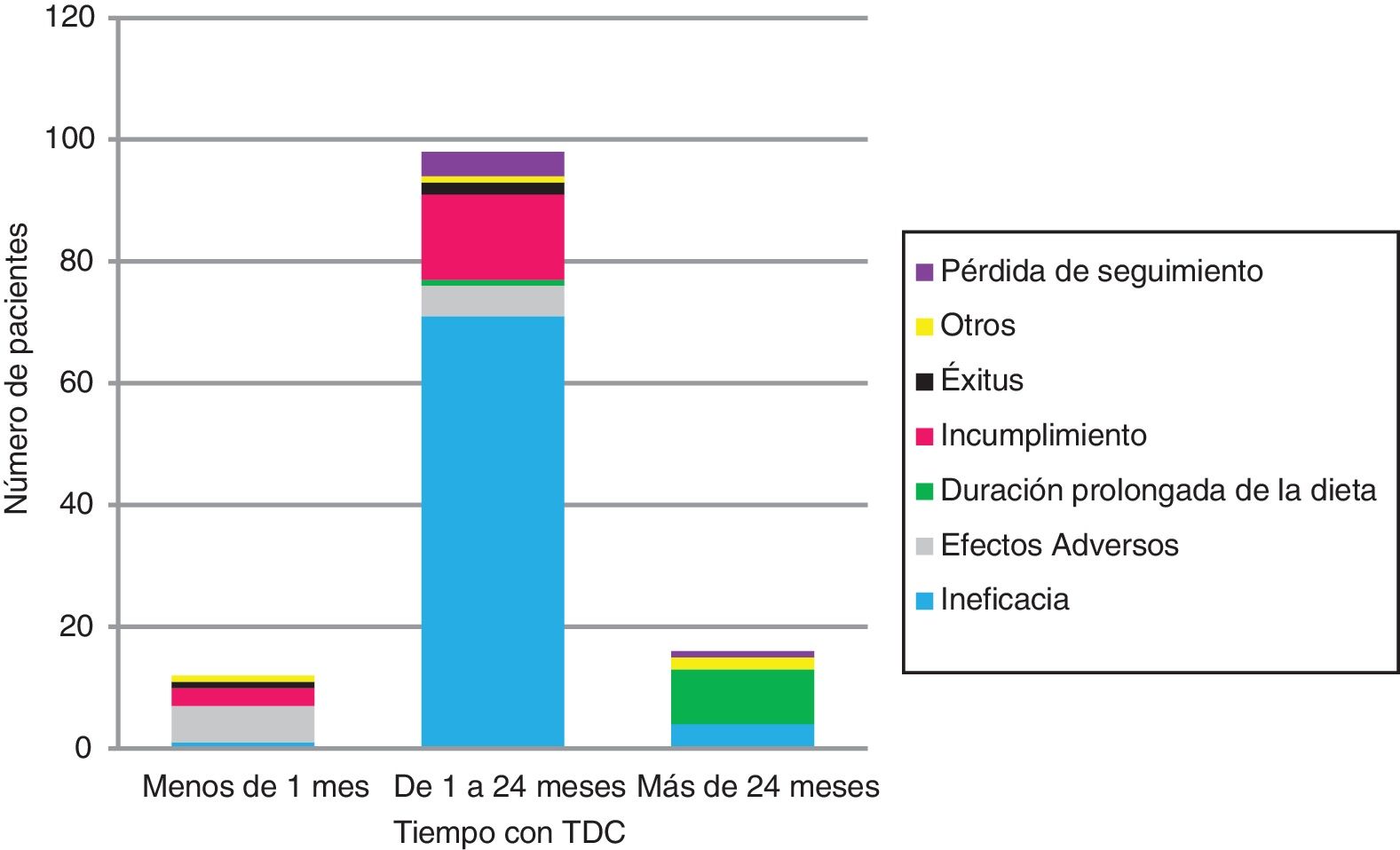
Three infants (2.4%) died while on the KD of causes unrelated to the dietary intervention. All were patients with encephalopathy managed with the CKD with a 3:1 ratio who experienced more than 10 seizures a day and were taking at least 4 AEM.
Side effects of ketogenic dietary teraphiesSeventy-seven patients (48%) had early AEs (Table 5). The most frequent AEs were gastrointestinal problems (25%), hypoglycaemia (18%; asymptomatic in 90%) and hypercalciuria (11%). Other AEs were dyslipidaemia (10), somnolence/asthenia (9; with hypoglycaemia in 2, with hyperammonaemia in 1 and with hyperketonaemia in 1), dehydration (4), acidosis (3) and anorexia (2). In 6 patients, AEs led to discontinuation of the diet: 2 on the MAD (due to asthenia and somnolence in 1 and to acidosis, associated with the use of topiramate, in the other), 3 on the CKD with a 3:1 ratio (due to vomiting, hyperammonaemia and hypertriglyceridaemia [associated with carnitine deficiency and supplementation with valproic acid] and recurrent urticaria associated with mild cow’s milk protein allergy) and 1 on the MCT-KD (due to vomiting and diarrhoea).
Early adverse events associated with ketogenic diets.
| Type of diet | Patients with early AEs, n (%)a |
|---|---|
| CKD, 3:1 ratio (n = 68) | 35 (51.5%) |
| CKD, 4:1 ratio (n = 16) | 11 (68.7%) |
| MAD (n = 58) | 23 (39.7%) |
| MCT-KD (n = 2) | 1 (50%) |
| Modified MCT-KD (n = 16) | 7 (43.7%) |
AE, adverse event; CKD, classic ketogenic diet; MAD, modified Atkins diet; MCT-KD, medium-chain triglyceride ketogenic diet.
Late AEs were frequent. Table 3 present the abnormal laboratory results and the percentage of patients that developed these alterations. Cholesterol levels were above baseline levels in every time interval (P ≤ .002). Triglyceride levels were elevated through 12 months (P ≤ .007). None of the patients required lipid-lowering drugs. The most frequent hepatic abnormality was elevation of gamma-glutamyl transpeptidase (GGT). However, the levels of GGT decreased significantly at 3 months (P = .008). Hypercalciuria was common, but only 1 patient developed nephrocalcinosis (0.62%). All patients with hypercalciuria received potassium citrate. Gastrointestinal problems were also common (10-15%), and the most frequent one was constipation, which was treated with polyethylene glycol.
In the MAD group, cholesterol levels were highest at 3 months. Triglyceride levels were highest in patients treated with the CKD and urea levels highest in patients treated with the MAD throughout the follow-up. Uric acid levels and the calcium/creatinine and protein/creatinine ratios in urine were higher in patients on the CKD at 6 months.
Nutritional assessment and anthropometric changesFig. 3 presents the anthropometric changes. The percentages of patients with a height z-score of less than –2 were: 14.4% at baseline, and 15.3%, 17.7%, 21.6% and 24.1% at the subsequent time points in the follow-up. Overall, we found significant differences in the height z-score at 12 months (–0.781 ± 1.66) compared to baseline (–0.312 ± 1.70) (P = .015) and at 24 months (–0.927 ± 1.20) compared to baseline (–0.310 ± 1.23) (P = .023). When we compared the impact on height in different age groups, we found a greater impact on younger children (Fig. 3B), although the differences were not statistically significant. Based on the type of diet, we found a greater decrease in the height z-score at 3 months (P = .009) and 12 months (P = .005) in patients treated with the CKD compared to other diets, but there were no differences in height or body mass index.
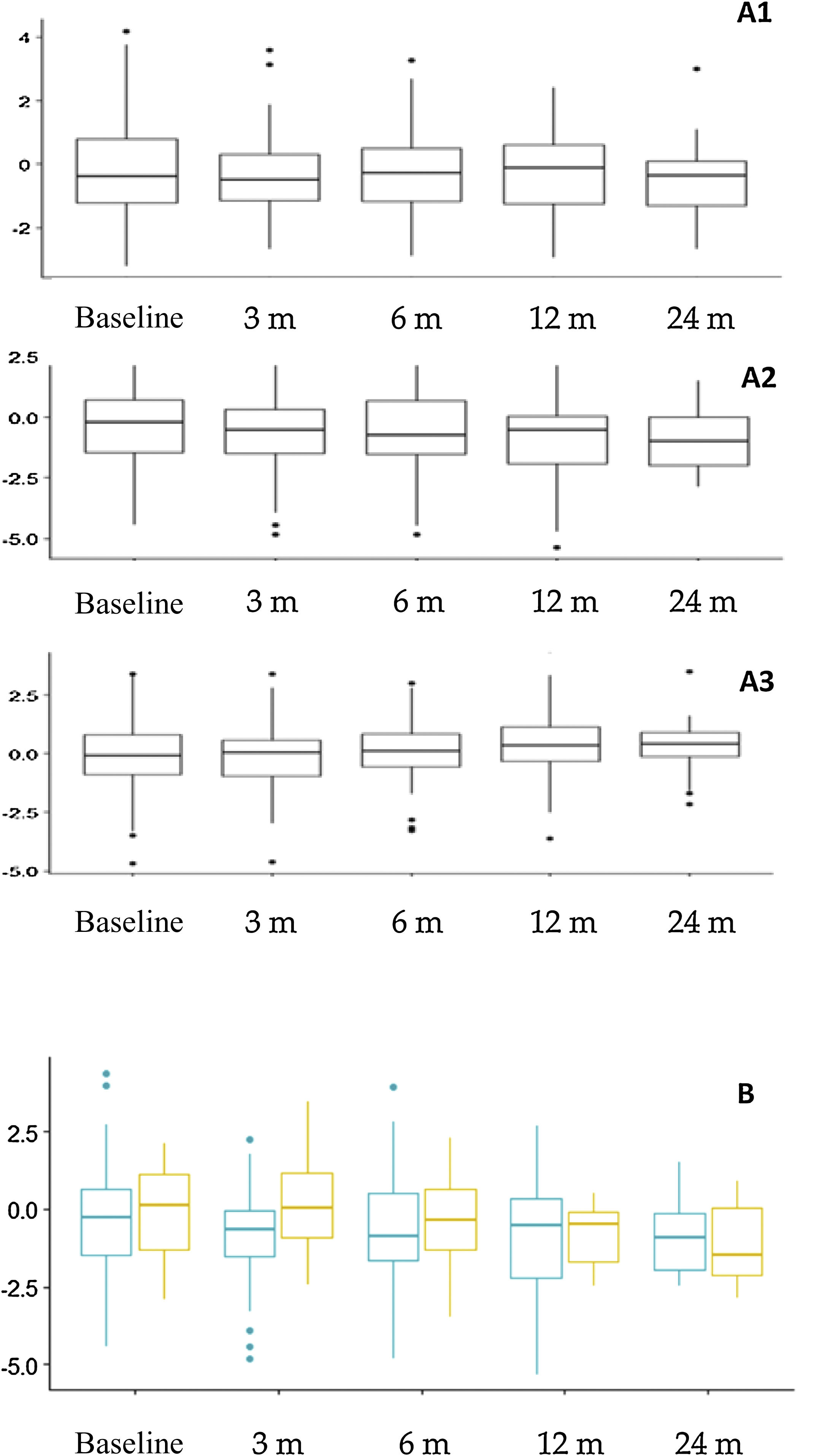
Anthropometric trends. A. Changes in z-scores for weight (A1), height (A2) and body mass index (A3) throughout the follow-up. B. Comparison of height z-score trends based at age at initiation of the ketogenic diet. Orange (right): patients that started the diet before age 2 years. Blue (left): patients that started the diet at or after age 2 years.
m, months.
We found increases in the levels of vitamin D (P = .05), folic acid (p < 0.001), ferritin (P = .009), selenium (P < .001) and carnitine (P = .008) at 12 months compared to baseline, and of ferritin (P = .047) and carnitine (P ≤ .02) at 24 months. There were decreases in the levels of prealbumin (P ≤ .034), retinol-binding protein (P ≤ .025) and magnesium (P ≤ .04) at 12 and 24 months, and of vitamin A (P = .03) at 12 months.
DiscussionOur study included the largest number of paediatric patients with epilepsy managed with KDT in Spain to describe the efficacy and safety of this approach. Previous studies found that 15% of patients became seizure-free and more than 30% achieved a reduction of 50% in the frequency of seizures.7,29–31 Previous studies have not found differences in efficacy based on age, the type of seizures or the type of KDT.4,7,8 In our cohort, 12% to 15% of patients became seizure-free and 41% to 16% exhibited a favourable response. We found that CKD was more effective, but only at 3 and 12 months of treatment. This tendency has been observed in randomised clinical trials, but the trend was not statistically significant.8
Although the KDT can be an efficacious intervention, ineffectiveness was the most frequent reason of discontinuation in our case series. Appropriate patient selection is important to the success of KDT. The KDT has proven more efficacious for management of the following32: GLUT1 deficiency syndrome, pyruvate dehydrogenase (PDH) deficiency, Doose, Ohtahara, Dravet and Angelman syndrome, complex 1 mitochondrial respiratory chain deficiency and tuberous sclerosis. One fourth of our patients had one of these diseases. Fifteen patients with these diseases were managed with a KDT for at least 2 years, and the reduction in the frequency of seizures was greater than 90% in every case. The proportion of patients that became seizure-free was highest in the group with seizures with a metabolic aetiology, probably because half of them had GLUT1 or PDH deficiency. The KDT is the first-line treatment for both disorders, in which brain metabolism is compromised, as ketones provide an alternative energy substrate. In patients with structural epilepsy, the KDT was also effective, but in fewer than half the cases. In this aetiological group, KDT interventions can serve as a temporizing measure until the patient can undergo surgery for definitive treatment, achieving an improvement, if only partial, in the seizures and sometimes a reduction in the use of ASM and their AEs.
Infants also benefit from KDT. We found that 16% to 28% became seizure-free and that 24% to 60% responded well to the intervention. Nordli et al33 reported similar outcomes, and a recent review concluded that KDT, contrary to what was thought in the past, are safe and effective for treatment of refractory epilepsy, GLUT1 deficiency syndrome and PDH deficiency in children aged less than 2 years.34 In this age group, early implementation of the KDT should be considered, as adherence is easier, and the CKD should be considered the first-line treatment.32
Adverse events were not a frequent cause of discontinuation (save in the first month) and were usually mild, transient and easy to manage. Gastrointestinal AEs were very prevalent, although less frequent compared to other studies that reported them in more than half the children.11,12 Hypercalciuria is a potential AE that is a source of concern, as it is asymptomatic and can result in severe complications such as nephrolithiasis. Hypercalciuria was very frequent, but the incidence of lithiasis in our patients was much lower compared to the previous literature.14 Ongoing monitoring of the urine calcium/creatinine ratio and early administration of potassium citrate after detection of hypercalciuria are probably the reasons for the low incidence found in our study. Dyslipidaemia was also frequent and developed early, as described in previous studies.15 The long-term cardiovascular risk is still under debate, and a previous study found normalization of the lipid profile at 12 months.15 Cholesterol levels were elevated throughout the follow-up, while the incidence of hypertriglyceridaemia in our sample decreased at 1 year. Dyslipidaemia could be controlled through dietary modification alone, replacing saturated fats (used to improve the palatability of the diet) by monounsaturated and polyunsaturated fats.
Dietary restrictions are another drawback of KDT. In fact, the second reason for discontinuation in our cohort was difficulty adhering to the diet. Interestingly, although the CKD is more restrictive, non-adherence was more frequent in patients on the MAD. Age may be at play in this paradoxical result. The CKD is usually prescribed to younger children whose diet is easier to control, as it is based on fluid formulations or mashed or blended foods. As the child grows, problems with adherence may emerge. This, combined with the fact that the CKD seems to be more effective in younger children,8 supports the indication of CKD for first-line treatment in children aged less than 2 years. However, once problems with adherence emerge, switching the classic KD or modified Atkins diet to less restrictive diets, such as the LGID, may help continue treatment.
The impact of KDT on nutritional status has been the subject of thorough study.16,17 The intake of calcium and vitamin D decreases in KDT. In addition, their levels tend to be low in children with severe epilepsy, and this can lead to serious adverse events such as fractures. In our cohort, the levels of selenium, carnitine and vitamin D, which tend to decrease with KDT, improved after initiation of the diet, as deficiencies were identified at baseline and the necessary supplements prescribed. Thus, we must underscore the importance of performing an adequate baseline assessment The impact on growth is still under debate. Growth delays may occur in 20% to 30% of patients,18–22,35 but catch-up growth can also occur after discontinuation of the diet.36 Long-term treatment with a KDT may have a significant impact on height, especially in the youngest patients.37 We found statistically significant differences in the height z-score during the follow-up, with values of less than –2 in 15% to 24% of our patients, with a higher frequency in children aged less than 2 years at the time of initiation of the dietary intervention. Some of the potential causes of faltering growth are inadequate energy and protein intake, ketosis,21,22 acidosis and endocrine changes.35 We did not find significant differences in growth based on the type of diet, and the protein intake was sufficient in every case. However, it is important to consider that children aged under 2 years followed a CKD, and it is precisely this subset of patients that is most vulnerable. It would be useful to identify patients at highest risk of growth faltering before initiation of a KDT (age < 2 years) and during treatment (acidosis, ketosis, endocrine changes) to monitor them more closely. Ketogenic dietary interventions usually last a minimum of 2 years. However, in the case of infantile spasms (a type of seizure that typically occurs during infancy), a previous study found no difference in the relapse rate between seizure-free patients that discontinued the diet at 8 months or at 2 years.38 Given the potential impact on growth, a shorter duration of the diet should be considered in patients at risk, especially infants.39
The mechanisms of action of ketogenic dietary interventions are still under investigation. Ketosis is among those studied most thoroughly. Although fewer than half of our patients achieved optimal levels of beta-hydroxybutyrate, most of them improved. This suggests that KDT have anticonvulsant and neuroprotective effects through other mechanisms.
There are limitations to our study. It had an observational and retrospective design, the number of patients varied between groups, and treatment protocols were not homogeneous throughout the study period. This limits extrapolation of the results to clinical practice, especially when it comes to the better outcomes observed with the CKD. In addition, it is not clear that the ketogenic dietary interventions were solely responsible for observed delays in growth. Epilepsy itself, underlying diseases and other treatments may also contribute to the impact on growth.
In conclusion, KDT are effective for management of refractory epilepsy in children. Adverse events are frequent but generally mild and easy to treat. Patients managed with a KDT require close follow-up by qualified medical staff to identify and treat AEs and monitor growth, which may be impacted, especially in younger children. In addition, ketogenic dietary therapies must be approached as a dynamic intervention susceptible of modification to reduce the incidence of AEs, improve adherence or attenuate the impact on nutritional status.
FundingThis research did not receive any external funding.
Conflicts of interestDr Jana Ruiz Herrero has participated in 2 courses on the ketogenic diet sponsored by Nutricia and has received funding for travel from Nutricia.
Dr Cañedo Villarroya has received fees and non-financial support from Nutricia, Mead Johnson, Nestlé, Abbott and Orphan.
Dr García Peñas has received fees from Nutricia, Sanofi, UCB, Eisai/Esteve, Bial and GW.
Ms Puerta Macfarland has participated in 2 courses on the ketogenic diet sponsored by Nutricia.
Dr Consuelo Pedrón Giner has served as a consultant and received fees for conferences from Nutricia, Vitaflo, Nestlé and Mead Johnson; has received funding for travel from Nutricia, Vitaflo and Nestlé.
Ms García Alcolea and Gómez Fernández have no conflicts of interest to disclose.
Please cite this article as: Ruiz Herrero J, Cañedo Villarroya E, García Peñas JJ, García Alcolea B, Gómez Fernández B, Puerta Macfarland LA, et al. Terapias dietéticas cetogénicas en epilepsia: experiencia en 160 pacientes durante 18 años. An Pediatr (Barc). 2022;96:511–522.
