



Las terapias dietéticas cetogénicas (TDC) tienen efecto neuroprotector y anticonvulsivante, reducen las crisis epilépticas y mejoran el estado cognitivo en pacientes epilépticos. Nuestro propósito fue evaluar los efectos de las TDC en niños con epilepsia refractaria (eficacia, efectos secundarios, impacto en el estado nutricional y crecimiento).
MétodosSe realizó un estudio observacional descriptivo retrospectivo y prospectivo en un hospital terciario español (enero de 2000-diciembre de 2018). Ciento sesenta pacientes pediátricos con epilepsia fueron tratados con TDC (82 varones; edad media 5 años 9 meses). Las convulsiones, los fármacos antiepilépticos, la antropometría, los efectos secundarios y los parámetros analíticos se controlaron al inicio del tratamiento y a los 3, 6, 12 y 24 meses.
ResultadosEn estos intervalos los pacientes libres de crisis fueron: 13,7%, 12,5%, 14,4% y 10,6%, respectivamente, lográndose una reducción de las convulsiones≥50% en el 41,9%, 37,5%, 28,7% y 16,2%. Los efectos secundarios fueron frecuentes, especialmente trastornos digestivos, hipercalciuria, hipoglucemia, disfunción hepática y dislipidemia. La prealbúmina, la proteína de unión al retinol, la vitamina A y el magnesio disminuyeron significativamente. La talla se vio afectada, especialmente en niños menores de 2 años.
ConclusionesLas TDC son efectivas para la epilepsia refractaria infantil. Sin embargo, los efectos adversos son frecuentes y pueden afectar al estado nutricional y al crecimiento.
Ketogenic dietary therapies (KDT) produce anticonvulsant and neuroprotective effects, reduce seizures and improve the cognitive state in patients with epilepsy. Our purpose was to evaluate the effects of KDT in children with refractory epilepsy (effectiveness, side effects, impact on nutritional status and growth).
MethodsA retrospective and prospective observational descriptive study was conducted in a Spanish tertiary hospital (January 2000 to December 2018). One hundred sixty pediatric patients with epilepsy were treated with KDT (82 males; mean age 5 years 9 months). Seizures, anti-epileptic drugs, anthropometric measures, side effects, and laboratory assessment were monitored baseline and at 3, 6, 12 and 24 months after the onset of KDT.
ResultsIn these time intervals, the seizure-free patients were: 13.7, 12.5, 14.4 and 10.6%, respectively, and a reduction of seizures≥50% was achieved in 41.9, 37.5, 28.7 and 16.2%. Side effects were frequent, especially digestive disorders, hypercalciuria, hypoglycemia, hepatic dysfunction and dyslipidemia. Prealbumin, retinol binding protein, vitamin A and magnesium decreased significantly. Height was affected, especially in children below 2 years.
ConclusionsKDT are effective for refractory epilepsy in children. However, adverse effects are frequent, and it may affect nutritional status and growth.
La epilepsia es uno de los principales trastornos neurológicos infantiles. Los fármacos antiepilépticos (FAE) mejoran las convulsiones en la mayoría de los pacientes. Sin embargo, hasta un tercio (7-20% de los niños y 30-40% de los adultos)1,2 pueden padecer epilepsia refractaria que puede asociarse con deterioro cognitivo, efectos adversos (EA) y mala calidad de vida. En estos casos las terapias dietéticas cetogénicas (TDC) son una alternativa terapéutica.
Existen diferentes tipos de TDC3. La dieta cetogénica clásica (DCC) proporciona un 87-90% de la ingesta calórica diaria como grasas3. La provisión de hidratos de carbono y proteínas varía en proporciones de 3:1 a 4:1 (3 a 4 gramos de grasa por 1 gramo de hidratos de carbono y proteínas). En la dieta cetogénica con triglicéridos de cadena media (TCM, DC-TCM) el 71% de la ingesta calórica total proviene de la grasa, siendo el 11% grasa natural y el 60% TCM4. La DC-MCT modificada permite un mayor contenido de grasas naturales (41%). La dieta de Atkins modificada (DAM) permite la ingesta libre de proteínas y grasas con restricción de hidratos de carbono (10gramos/día)5. La dieta de bajo índice glucémico (DBIG) liberaliza la toma de hidratos de carbono (40-60gramos/día), favoreciendo aquellos con índice glucémico<506. No existe evidencia clara de diferencias de eficacia entre DCC, DC-TCM7 o DAM8. Algunos estudios sugieren que la DBIG también es eficaz9, a pesar de no inducir cetosis, y en un ensayo clínico aleatorizado reciente no fue inferior a la DCC10.
Las TDC pueden causar problemas digestivos11,12, hepáticos12, renales13,14, dislipidemia15, déficits nutricionales16,17 y alteraciones del crecimiento18–22.
Los objetivos de este estudio fueron evaluar la eficacia de las TDC, sus EA y el impacto sobre el crecimiento y el estado nutricional. Se compararon diferentes tipos de TDC y grupos de edad para identificar diferencias en los ítems estudiados.
Pacientes y métodosSe recogieron de forma anónima los datos de todos los pacientes<18 años con epilepsia tratados con TDC en un hospital terciario entre enero de 2000 y diciembre de 2018. Es un estudio observacional descriptivo retrospectivo y prospectivo (desde mayo de 2015) aprobado por el Comité de Ética en Investigación Clínica del Hospital Infantil Universitario Niño Jesús (R-0002/15). Los pacientes (o sus familiares) fueron informados de la investigación, firmaron un consentimiento informado y se sometieron a un examen basal y a los 3, 6, 12 y 24 meses.
Para la calibración de las dietas las recomendaciones energéticas de la Organización Mundial de la Salud (OMS)23, según edad y peso, se utilizaron cuando no se realizó una calorimetría indirecta. La ingesta proteica se calculó según las recomendaciones (1g/kg/día en>1 año y 1,5 en lactantes)24. El método de Holliday-Segar25 se empleó para estimar los requerimientos mínimos de líquidos, aunque se prescribió al menos la recomendación de la OMS26. Se pautaron suplementos de oligoelementos, vitaminas y minerales sin hidratos de carbono para alcanzar al menos la ingesta diaria recomendada26,27 cuando se encontraron aportes insuficientes o deficiencias.
La eficacia se evaluó según la reducción del número de FAE y el porcentaje de reducción de crisis en comparación con el valor inicial: 100% (libre de crisis), 90-100%, 50-90%, <50%, 0% (sin mejoría) o empeoramiento.
Los EA se dividieron en precoces (primer mes) o tardíos. Se analizaron peso, talla e índice de masa corporal y se ajustaron por sexo y edad según las tablas de la OMS. La talla se consideró normal cuando la puntuación z estaba entre –2 y+2, alteración leve si z era<–2 y≥−3 y alteración grave si z era<−3. La evaluación de laboratorio incluyó hemograma, perfil bioquímico y lipídico, gasometría, sedimento urinario y cocientes calcio, proteína y citrato/creatinina, prealbúmina, proteína transportadora de retinol, vitaminas A, D, E, B9, B12, cinc, selenio, paratohormona y carnitina.
Los datos se analizaron con el software SPSS Statistics versión 16.0. Los datos cuantitativos se presentan como media y desviación estándar (DE) para distribuciones normales, y mediana y rango o rango intercuartílico en caso contrario. Los datos cualitativos se expresan como porcentaje. Se realizó un análisis por intención de tratar.
Se comparó la eficacia de 3 grupos de dieta: DCC (relación 3:1 y 4:1), DC-TCM (clásica o modificada) y DAM. Para detectar diferencias entre los parámetros antropométricos basalmente y en diferentes intervalos de seguimiento según el tipo de TDC y grupo de edad (<2 años, 2-5 años, 5-10 años y>10años) se aplicó el modelo de análisis de varianza de medidas repetidas (ANOVA) para datos apareados. Se utilizó el test exacto de Fisher para comparar la eficacia entre diferentes TDC. Se empleó la «t» de Student para datos apareados para comparar los valores analíticos basales y a los 12 y 24 meses. Se utilizaron ANOVA y la «t» de Student para muestras independientes para comparar dichos indicadores según el tipo de TDC. Se consideraron significativos valores de p≤0,05.
ResultadosSe trataron 160 pacientes con TDC, 71 desde mayo de 2015. Sus características se especifican en la tabla 1. Todos padecían epilepsia refractaria, excepto un paciente que no había recibido FAE por sospecha de síndrome de deficiencia del transportador cerebral de la glucosa tipo 1 (SD-GLUT1). La tabla 2 muestra las causas de la epilepsia. La mediana de edad al inicio de la epilepsia fue de 291 días (rango intercuartílico=936,2). El 90% tenía convulsiones diariamente. El promedio de FAE utilizados antes de la TDC fue de 6 (rango 0-16), y el 59% presentó EA. Otros tratamientos utilizados fueron: estimulador del nervio vago (5), cirugía (7), corticoides (48), gammaglobulina (15) y otros como vitamina B6 (47%). Doce recibieron previamente TDC, con buena respuesta en 5.
Características de los pacientes del estudio y de las terapias dietéticas cetogénicas
| Pacientes con TDC (n = 160) | |
|---|---|
| Género | Varón, 82 (51%)Mujer, 78 (49%) |
| Retraso psicomotor/discapacidad intelectual | 73% |
| Tipo de crisis | Tónico-clónica generalizada, 10 (6,2%)Mioclónica, 27 (16,9%)Tónica, 30 (18,7%)Focal, 37 (23%)Atónica, 3 (1,9%)Ausencia, 7 (4,4%)Clónica, 6 (3,7%)Varios, 40 (25%) |
| Edad al inicio de la epilepsia | Recién nacido, 21 (13,1%)Lactante, 68 (42,5%)1 a 5 años, 54 (33,7%)5 a 10 años, 12 (7,5%)>10años, 5 (3,1%) |
| Número de crisis antes del inicio de la TDC | ≤1/día, 16 (10%)2-10/día, 89 (55,6%)>10/día, 35 (21,9%)Incontables, 3 (1,9%)Desconocido, 5 (3,1%)Estatus epiléptico, 12 (7,5%) |
| Edad al inicio de la TDC | Lactante, 25 (15,6%)1 a 2 años, 15 (9,4%)2 a 5 años, 45 (28,1%)5 a 10 años, 40 (25%)>10 años, 35 (21,9%) |
| Tipo de TDC | DCC, 3:1; 68 (42,5%)DCC, 4:1; 16 (10%)DAM, 58 (36,2%)DC-TCM, 2 (1,2%)DC-TCM, modificada, 16 (10%) |
| Vía de administración | Oral, 120 (75%)SNG, 24 (15%)Gastrostomía, 16 (10%) |
| Mediana de duración de la TDC (rango) | 259 días (5 días-9 años y 6 meses) |
| Motivos de retirada de la TDCa | Ineficacia, 76 (60,3%)Incumplimiento, 17 (13,5%)Efectos adversos, 11 (8,7%)Duración >2años, 10 (8%)Pérdida de seguimiento, 5 (4%)Fallecimiento, 3 (2,5%)Otras, 4 (3%) |
DAM: dieta de Atkins modificada; DCC: dieta cetogénica clásica; DC-TCM: dieta cetogénica con triglicéridos de cadena media; SNG: sonda nasogástrica; TDC: terapia dietética cetogénica.
Etiología de la epilepsia
| Etiología | Frecuencia (%) | Causa específica |
|---|---|---|
| Desconocida | 29 (18,1) | |
| Metabólica | 24 (15) | 11 SD-GLUT1 (5 confirmados genéticamente)9 enfermedad mitocondrial2 lipofuscinosis neuronal ceroidea (conocidas a posteriori)Una hipoglucemia neonatal graveUn déficit de PDH |
| Estructural | 43 (26,9) | 22 malformaciones del desarrollo cortical (15 displasia, 3 lisencefalia, 2 polimicrogiria, una hemimegalencefalia, una heterotopia subcortical)16 daños cerebrales hipóxico-isquémicos (un déficit de proteína C, una mutación del factor viii, una meningitis, una sepsis, una leucemia, 9 enfermedades perinatales, un infarto cerebral, una complicación de cirugía cardíaca compleja)2 túberes corticales (complejo esclerosis tuberosa)Una malformación cerebral compleja (megalencefalia y anomalía de migración neuronal hemisférica [polimicrogiria y heterotopias nodulares periventriculares]) y quiste aracnoideo giganteUna agenesia de cuerpo calloso |
| Un angioma leptomeníngeo (Sturge Weber) | ||
| Genética | 39 (24,4) | 28 mutaciones genéticas (6 CDKL5; 4 SCN1A [síndrome de Dravet]; 3 KCNQ2; 3 STXBP1; 2 SLC9; 2 MECP2 ligado a X; un SCN2A (encefalopatía epiléptica precoz); un CHRNA4 (epilepsia nocturna del lóbulo temporal); un SCN1B; un RBFOX1; un CACNA1H; un CNTNAP2 (asociado a displasia y síndrome Pitt Hopkins like); un PTPN11 (síndrome de Noonan); un PHOX2B (síndrome de Ondine)11 trastornos cromosómicos (3 trisomía 21 [síndrome de Down]; 3 inversión-duplicación de la región proximal del cromosoma 15; una deleción 1p; una deleción 11q14; un síndrome de Angelman; un cromosoma 17 en anillo; una duplicación 16p) |
| Infecciosa | 6 (3,7) | 3 Virus herpes simple |
| 2 Streptococcus pneumoniaeUn citomegalovirus (perinatal) | ||
| Inmune | 4 (2,5) | 2 confirmadas (anticuerpos antiNMDA positivos) |
| Síndromes | 15 (9,4) | 3 síndrome de Lennox-Gastaut3 síndrome de West3 FIRES4 síndrome de DooseUn síndrome de OhtaharaUn síndrome de Landau Kleffner |
CACNA1H: calcium voltage-gated channel subunit alpha 1H; CDKL5: cyclin dependent kinase-like 5; CHRNA4: cholinergic receptor nicotinic alpha 4 subunit; CNTNAP2: contactin associated protein like 2; FIRES: síndrome epiléptico relacionado con infección febril; KCNQ2: potassium voltage-gated channel subfamily Q member 2; MECP2: methyl-CpG binding protein 2; NMDA: N-methyl-D-aspartate; PDH: piruvato deshidrogenasa; PHOX2B: paired like homeobox 2B; RBFOX1: RNA binding fox-1 homolog 1; SCN1A: sodium voltage-gated channel alpha subunit 1; SCN2A: sodium voltage-gated channel alpha subunit 2; SCN1B: sodium voltage-gated channel beta subunit 1; SD-GLUT1: síndrome de deficiencia del transportador cerebral de la glucosa tipo 1; SLC9: solute carrier family 9; STXBP1: syntaxin binding protein 1; PTPN11: protein tyrosine phosphatase non-receptor type 11.
La edad media al inicio de la TDC fue de 5 años y 9 meses (rango: 79 días-17 años y 4 meses). El tiempo medio desde el inicio de los síntomas hasta el tratamiento con TDC fue de 3 años y 10 meses (rango 7 días-14 años y 6 meses).
Antes de la TDC se encontraron algunas alteraciones en marcadores nutricionales (tabla 3), y el 16,8% presentaba una ingesta deficiente de vitaminas y oligoelementos.
Evolución de parámetros de laboratorio en sangre y orina
| Valores de referencia | Media (DE) | Número de pacientes por encima o por debajo del valor de referencia (%)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Intervalo de tiempo(número de pacientes) | Basal(160) | 3m(126) | 6m(95) | 12m(62) | 24m(32) | Basal | 3m | 6m | 12m | 24m | |
| Marcadores nutricionales en sangre | |||||||||||
| Prealbúmina (mg/dl) | 15-40 | 21,3 (6,5) | 18,5 (4,7) | 18,4 (4,5) | 19 (6,2) | 17,8 (3,8) | 16 (10) | 21 (16,6) | 19 (20) | 15 (24,2) | 7 (21,9) |
| RBP (mg/dl) | 2,5-6,9 | 3,5 (1,2) | 2,6 (1,3) | 2,6 (1,1) | 2,7 (1,0) | 2,6 (0,7) | 32 (20) | 20 (15,9) | 22 (23,2) | 20 (32,3) | 7 (21,9) |
| Ferritina (ng/ml) | 7-140 (<15a)15-175 (≥15a) | 46,8 (57) | 42,6 (43) | 51,9 (44) | 58,4 (37) | 55,3 (39,7) | 4 (2,5) | 3 (2,4) | 0 | 0 | 1 (3) |
| Micronutrientes | |||||||||||
| Magnesio (mg/dl) | 1,5-2,5 | 2,1 (0,3) | 2,0 (0,2) | 2,1 (0,2) | 2,0 (0,2) | 1,97 (0,1) | 1 (0,6) | 1 (0,8) | 0 | 0 | 0 |
| Selenio (μg/l) | 70-120 | 71,2 (21) | - | 87,7 (14) | 85,7 (21) | 87,5 (14,5) | 31 (19,3) | - | 2 (2,1) | 4 (6,4) | 1 (3) |
| Cinc (μg/dl) | 70-150 | 91,2 (32) | - | 102,5 (24) | 95,7 (24) | 101,4 (17,3) | 12 (7,5) | - | 2 (2,1) | 4 (6,4) | 0 |
| Carnitina total o levocarnitina (μmol/l) | 21,5-64,520-50 | 55,6 (25)42,1(18) | -- | 84,8 (47)47,2 (24) | 81,2 (40)46,5 (18) | 69 (31,4)47 (16,7) | 17 (10,6) | - | 2 (2,1) | 5 (8) | 1 (3) |
| Vitamina A (mg/l) | 0,2-0,6 | 0,43 (0,2) | 0,37 (0,1) | 0,35 (0,1) | 0,35 (0,1) | 0,36 (0,2) | 3 (1,9) | 1 (0,8) | 1 (1) | 4 (6,4) | 1 (3) |
| Vitamina E (mg/l) | 3-9 (<12a)5-20 (≥12a) | 5,6 (3,9) | 4,6 (1,8) | 5,1 (1,2) | 4,8 (1,4) | 5,0 (1,2) | 9 (5,6) | 3 (2,4) | 4 (4,2) | 6 (9,7) | 2 (6,2) |
| Ácido fólico (B9) (ng/ml) | 3,9-23,9 | 12,1 (6,2) | 16,6 (11) | 16,9 (5,8) | 19,1 (6,4) | 17 (5,4) | 5 (3,1) | 0 | 0 | 0 | 0 |
| Vitamina B12 (pg/ml) | 250-914 | 814 (624) | 955 (559) | 876 (355) | 897 (533) | 916 (489) | 3 (1,9) | 0 | 0 | 0 | 0 |
| 25-hidroxi-vitamina D (ng/ml) | 20-80 | 28,6 (11) | 35,8 (12) | 37,8 (14) | 34,4 (14) | 31 (10) | 25 (15,6) | 2 (1,6) | 3 (3,2) | 5 (8) | 3 (9,4) |
| Calcio (mg/dl) | 8,8-10,8 | 9,6 (0,4) | 9,6 (0,4) | 9,6 (0,4) | 9,6 (0,4) | 9,6 (0,3) | 3 (1,9) | 0 | 1 (1) | 0 | 0 |
| Fósforo (mg/dl) | 4,5-6,5 | 5,0 (0,8) | 4,9 (0,7) | 4,9 (0,7) | 4,7 (0,7) | 4,8 (0,5) | 31 (19,4) | 24 (19,0) | 20 (21) | 24 (38,7) | 9 (28) |
| Perfil lipídico | |||||||||||
| Colesterol (mg/dl) | 100-200 | 164 (37) | 195 (61) | 197 (53) | 196 (53) | 195 (44) | 23 (14,4) | 39 (31) | 32 (33,7) | 27 (43,5) | 14 (43,7) |
| Triglicéridos (mg/dl) | 35-115 | 81 (50) | 117 (87) | 108 (61) | 115 (95) | 88 (60) | 27 (16,9) | 27 (21,4) | 20 (21) | 10 (16,1) | 3 (9,4) |
| Marcadores hepáticos | |||||||||||
| AST (U/l) | 28-50 | 33,2 (16) | 30,1 (14) | 31,7 (17) | 33,7 (35) | 27,7 (6,7) | 16 (10) | 7 (5,5) | 6 (6,3) | 5 (8) | 0 |
| ALT (U/l) | 15-50 | 19,3 (16) | 20,6 (14) | 20,5 (13) | 23,7 (38) | 19 (8,2) | 4 (2,5) | 3 (2,4) | 4 (4,2) | 2 (3,2) | 0 |
| GGT (U/l) | 8-30 | 52,9 (110) | 33,7 (65) | 31,5 (56) | 27,9 (23) | 27 (24,4) | 56 (35) | 18 (14,3) | 12 (12,6) | 13 (21) | 6 (18,7) |
| Gasometría | |||||||||||
| pH | 7,35-7,45 | 7,4 (0,04) | 7,3 (0,03) | 7,3 (0,04) | 7,3 (0,04) | 7,3 (0,04) | 6 (3,7) | 7 (5,5) | 5 (5,3) | 5 (8) | 3 (9,4) |
| Bicarbonato (mmol/l) | 21-29 | 24,4 (6,5) | 22,3 (3,5) | 27,7 (7,9) | 21,9 (3,8) | 21,5 (2,9) | 12 (7,5) | 24 (19,0) | 8 (8,4) | 13 (21) | 7 (21,9) |
| Hormonas | |||||||||||
| IGF-1 (ng/ml) | 49-288 | 108 (77) | - | 117 (110) | - | 81,7 (49,5) | 10 (6,2) | - | 9 (9,5) | - | 4 (12,5) |
| PTH (pg/ml) | 15-80 | 38,9 (22) | 27,0 (11) | 32,4 (14) | - | 30,6 (12,2) | 6 (3,7) | 2 (1,6) | 4 (4,2) | 3 (4,8) | 2 (6,2) |
| Marcadores urinarios | |||||||||||
| Calcio/creatinina (mg/mg) | ≤0,8 (0-6 m)≤0,6 (6-12m)≤0,2 (≥1a) | 0,16 (0,17) | 0,3 (0,25) | 0,26 (0,24) | 0,27 (0,33) | 0,29 (0,25) | 19 (11,9) | 40 (31,7) | 27 (28,4) | 16 (25,8) | 15 (46,9) |
| Proteína/creatinina (mg/mg) | ≤0,5 (<2 a)≤0,2 (≥2 a) | 0,24 (0,15) | 0,33 (0,19) | 0,24 (0,16) | 0,31 (0,44) | 0,23 (0,28) | 12 (7,5) | 6 (4,8) | 5 (5,3) | 4 (6,4) | 6 (18,7) |
| Citrato/creatinina (mg/mg) | ≥0,4 | 0,8 (0,63) | 1,3 (2,7) | 0,8 (0,7) | 0,9 (0,9) | 0,6 (0,35) | 12 (7,5) | 9 (7,1) | 7 (7,4) | 11 (17,7) | 4 (12,5) |
| Cuerpos cetónicos | |||||||||||
| Beta-hidroxibutirato (mmol/l) | >2,4 | - | 3,1 (1,8) | 3,0 (1,7) | 3,5 (1,7) | 3,0 (1,7) | - | 47 (37,3) | 29 (30,5) | 27 (43,5) | 15 (46,9) |
a: años; ALT: alanino aminotransferasa; AST: aspartato aminotransferasa; GGT: gamma glutamil transpeptidasa; m: meses; IGF-1: factor de crecimiento insulínico tipo 1; PTH: hormona paratiroidea; RBP: proteína transportadora de retinol.
La tabla 1 muestra los tipos de dieta utilizados. Entre 2009 y 2014 se siguieron las recomendaciones del Grupo Internacional para el Estudio de la Dieta Cetogénica28. Desde 2015 se utilizó el protocolo del propio departamento de nutrición. La DCC se indicó en pacientes con sonda nasogástrica (SNG) o gastrostomía,<2 años, o en estatus. Se seleccionó la DAM para niños>2años. Las dietas se introdujeron aumentando progresivamente la ratio cetogénica, adecuándose a la tolerancia y los EA, sin restricción de líquidos.
Todos los pacientes<2 años (excepto 2) comenzaron DCC, ratio 3:1. Las DAM y DC-TCM solo se pautaron en>2 años. Se administró DBIG en 4 casos para mejorar el cumplimiento. Todas las DAM se administraron por vía oral. Todas las dietas administradas por sonda fueron DCC (excepto una DC-TCM modificada). El 25% comenzó en régimen ambulatorio (todas DAM). Se utilizó el ayuno en 15 (12 DC-TCM que comenzaron en 2006 o antes, y 3 DCC en epilepsia grave). Las calorías se restringieron en 4 niños que comenzaron antes de 2014 (75-83% de las recomendaciones diarias). El tiempo medio para alcanzar cetosis (>2,4μmol/l) fue 4,3 días (DE: 3,9; rango 12horas-30días). El beta-hidroxibutirato fue significativamente mayor en las DCC durante todo el seguimiento (excepto a los 6 meses).
Eficacia de las terapias dietéticas cetogénicasLa eficacia global se muestra en la tabla 4, y por grupos de edad en la figura 1. Cuatro lactantes no completaron 3 meses (2 por ineficacia y 2 fallecieron).
Eficacia de las terapias dietéticas cetogénicas
| Eficaciaa | 3 meses(n=126) | 6 meses(n=95) | 12 meses(n=62) | 24 meses(n=32) |
|---|---|---|---|---|
| Total de pacientes respondedores | 67 (41,9%) | 60 (37,5%) | 46 (28,7%) | 26 (16,2%) |
| Buena respuesta | 31 (19,4%) | 24 (15%) | 13 (8%) | 7 (4,4%) |
| Respuesta excelente | 14 (8,7%) | 16 (10%) | 10 (6,2%) | 2 (1,2%) |
| Libres de crisis | 22 (13,7%) | 20 (12,5%) | 23 (14,4%) | 17 (10,6%) |
La eficacia de la dieta se calculó comparando el número de crisis en cada periodo de seguimiento con las crisis antes de comenzar la dieta. Se consideraron respondedores los pacientes que presentaron una reducción de las crisis≥50%. Se consideró buena respuesta si la reducción fue del 50-90%, respuesta excelente si la reducción era>90% y libres de crisis los que no habían tenido ninguna crisis epiléptica. Los porcentajes se han calculado por intención de tratar teniendo en cuenta el total de la muestra (160 pacientes).
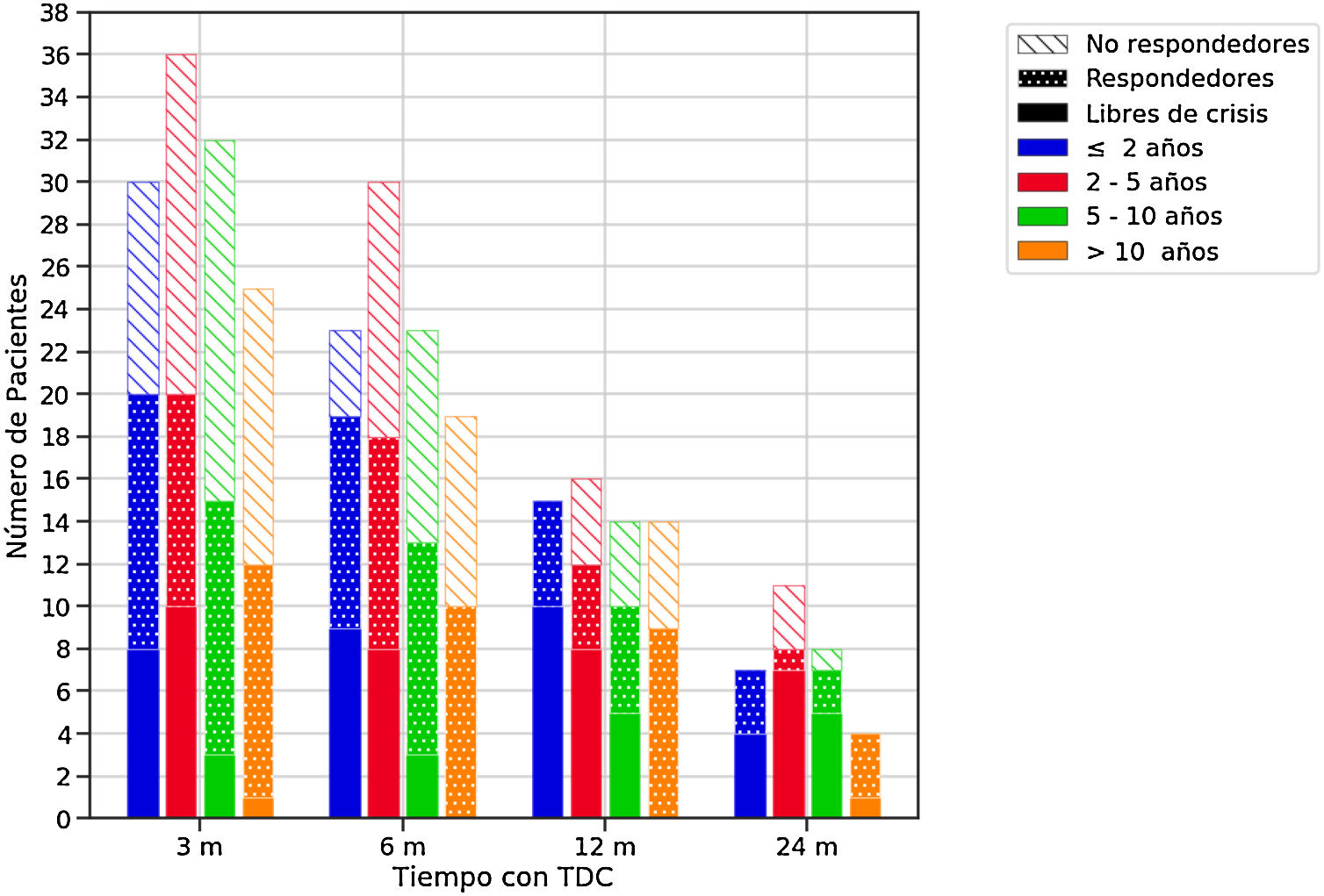
Eficacia de las terapias dietéticas cetogénicas (TDC) a lo largo del seguimiento en función de la edad al inicio de la dieta.
El color de las barras hace referencia a la edad de los pacientes al inicio de la dieta y el relleno a la respuesta. Se consideraron respondedores los pacientes que lograron una reducción de crisis de al menos un 50% con respecto al estado basal.
m: Meses.
La DCC fue más eficaz a los 3 y 12 meses (p<0,05). Según las diferentes etiologías en vez de las TDC fueron eficaces en el 45-10% de las epilepsias estructurales, en el 79-47% de las enfermedades metabólicas y en el 59-19% de los trastornos genéticos, a lo largo del seguimiento. Los FAE se redujeron en 30, 13, 9 y 9 niños en los diferentes intervalos. El porcentaje de pacientes que tomaba≤2FAE aumentó de un 34% antes de la dieta hasta un 41-56% a lo largo del seguimiento.
Se suspendió la TDC en 126 (tabla 1 y fig. 2). La ineficacia fue la primera causa de abandono en todos los grupos de edad y tipos de TDC, seguida del incumplimiento en la DAM (17/12) y los EA en la DCC (6/11). Ningún niño<2años abandonó la dieta por incumplimiento.
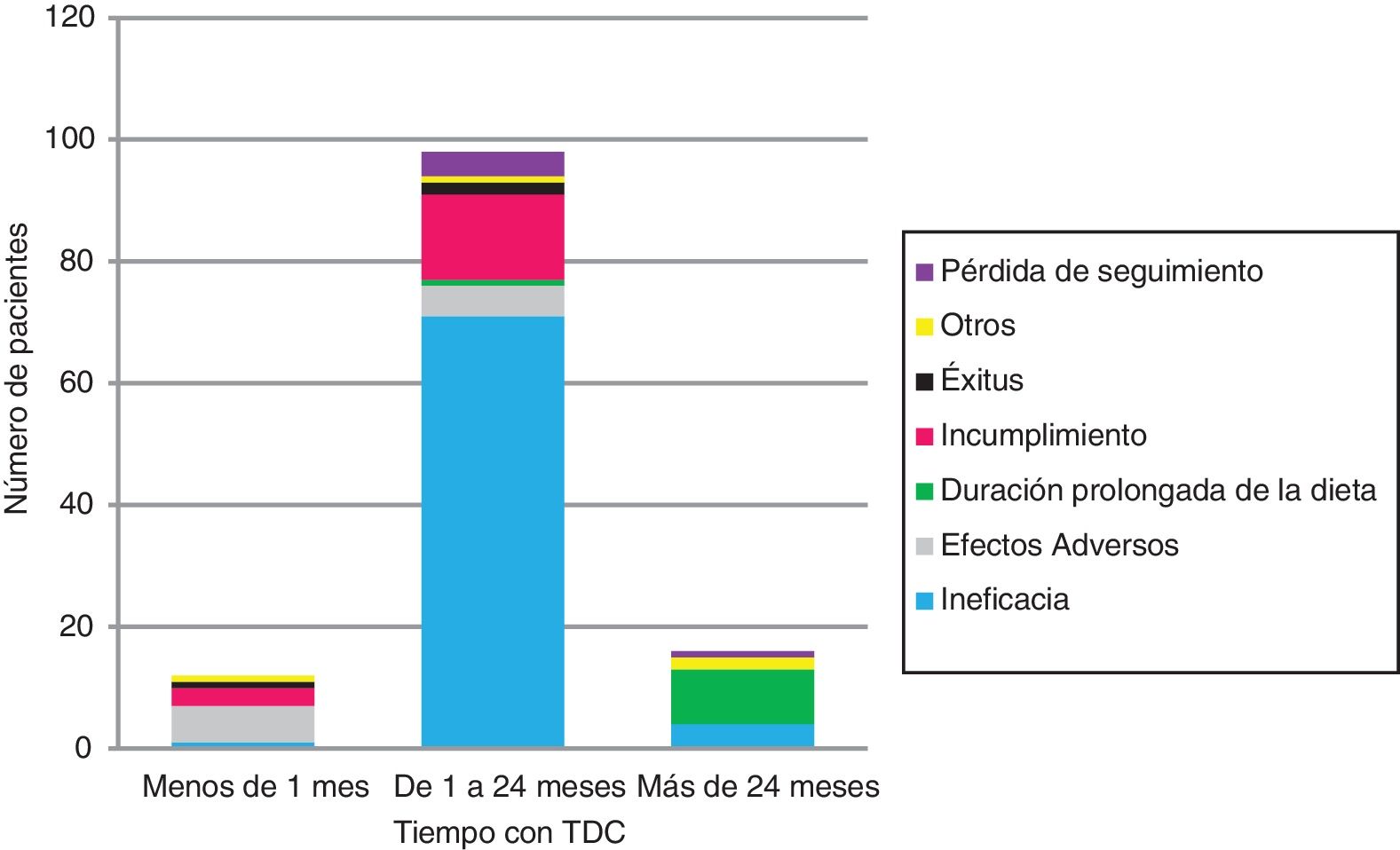
Tres lactantes (2,4%) murieron durante la terapia, sin relación con ella. Todos eran niños con encefalopatía con DCC ratio 3:1, con>10convulsiones/día y al menos 4 FAE.
Efectos secundarios de las terapias dietéticas cetogénicasSetenta y siete pacientes (48%) presentaron EA precoces (tabla 5). Los más frecuentes fueron: problemas digestivos (25%), hipoglucemia (18%; 90% asintomática) e hipercalciuria (11%). Otros fueron: dislipidemia (10), somnolencia/decaimiento (9; 2 con hipoglucemia, uno con hiperamonemia y uno con hipercetonemia), deshidratación (4), acidosis (3) y anorexia (2). En 6 sujetos los EA precoces conllevaron la retirada de la dieta: 2 DAM (por decaimiento y somnolencia, y por acidosis —relacionada con topiramato— respectivamente), 3 DCC ratio 3:1 (por vómitos, hiperamonemia e hipertrigliceridemia —relacionada con deficiencia de carnitina y toma de valproico— y urticaria recurrente relacionada con alergia leve a proteínas de leche de vaca) y una DC-MCT (por vómitos y diarrea).
Efectos adversos precoces de las terapias dietéticas cetogénicas
| Tipo de dieta | Número de pacientes con efectos adversos precoces (%)a |
|---|---|
| DCC ratio 3:1 (n=68) | 35 (51,5) |
| DCC ratio 4:1 (n=16) | 11 (68,7) |
| DAM (n=58) | 23 (39,7) |
| DC-TCM (n=2) | 1 (50) |
| DC-TCM modificada (n=16) | 7 (43,7) |
DAM: dieta de Atkins modificada; DCC: dieta cetogénica clásica; DC-TCM: dieta cetogénica con triglicéridos de cadena media.
Los EA tardíos fueron frecuentes. En la tabla 3 se presentan los parámetros analíticos que se alteraron y el porcentaje de pacientes que sufrió dichas alteraciones. El colesterol fue superior al valor basal en todos los intervalos de tiempo (p≤0,002). La hipertrigliceridemia se produjo hasta los 12 meses (p≤0,007). Ningún paciente precisó fármacos hipolipidemiantes. La alteración hepática más frecuente fue la elevación de la gammaglutamiltranspeptidasa (GGT). Sin embargo, esta se redujo significativamente a los 3 meses (p=0,008). La hipercalciuria fue común, pero solo un niño tuvo nefrocalcinosis (0,62%). Todos los individuos con hipercalciuria fueron tratados con citrato potásico. Los problemas gastrointestinales fueron frecuentes (10-15%), siendo el más común el estreñimiento, que se trató con polietilenglicol.
Los niveles de colesterol fueron superiores en las DAM a los 3 meses. Los triglicéridos fueron superiores en las DCC y la urea en las DAM durante todo el seguimiento. El valor del ácido úrico y las relaciones calcio/creatinina y proteína/creatinina urinarias fueron superiores en las DCC a los 6 meses.
Evolución nutricional y cambios antropométricosLa figura 3 muestra los cambios antropométricos. El porcentaje de pacientes con un z de talla<–2 fue: 14,4% basalmente y 15,3%, 17,7%, 21,6% y 24,1% en los diferentes intervalos de seguimiento. De forma global se encontraron diferencias significativas en el z de talla a los 12 meses (–0,781±1,66) en comparación con el valor basal (–0,312±1,70) (p=0,015) y a los 24 meses (–0,927±1,20) en comparación con el valor basal (–0,310±1,23) (p=0,023). Al comparar la repercusión sobre la talla en los distintos grupos de edad se observó un mayor impacto en los niños más pequeños (fig. 3B), aunque las diferencias no fueron estadísticamente significativas. En función del tipo de dieta se observó una disminución en la puntuación z del peso a los 3 meses (p=0,009) y 12 meses (p=0,005) en pacientes con DCC en comparación con las otras dietas, pero no hubo diferencias en la talla ni en el índice de masa corporal.
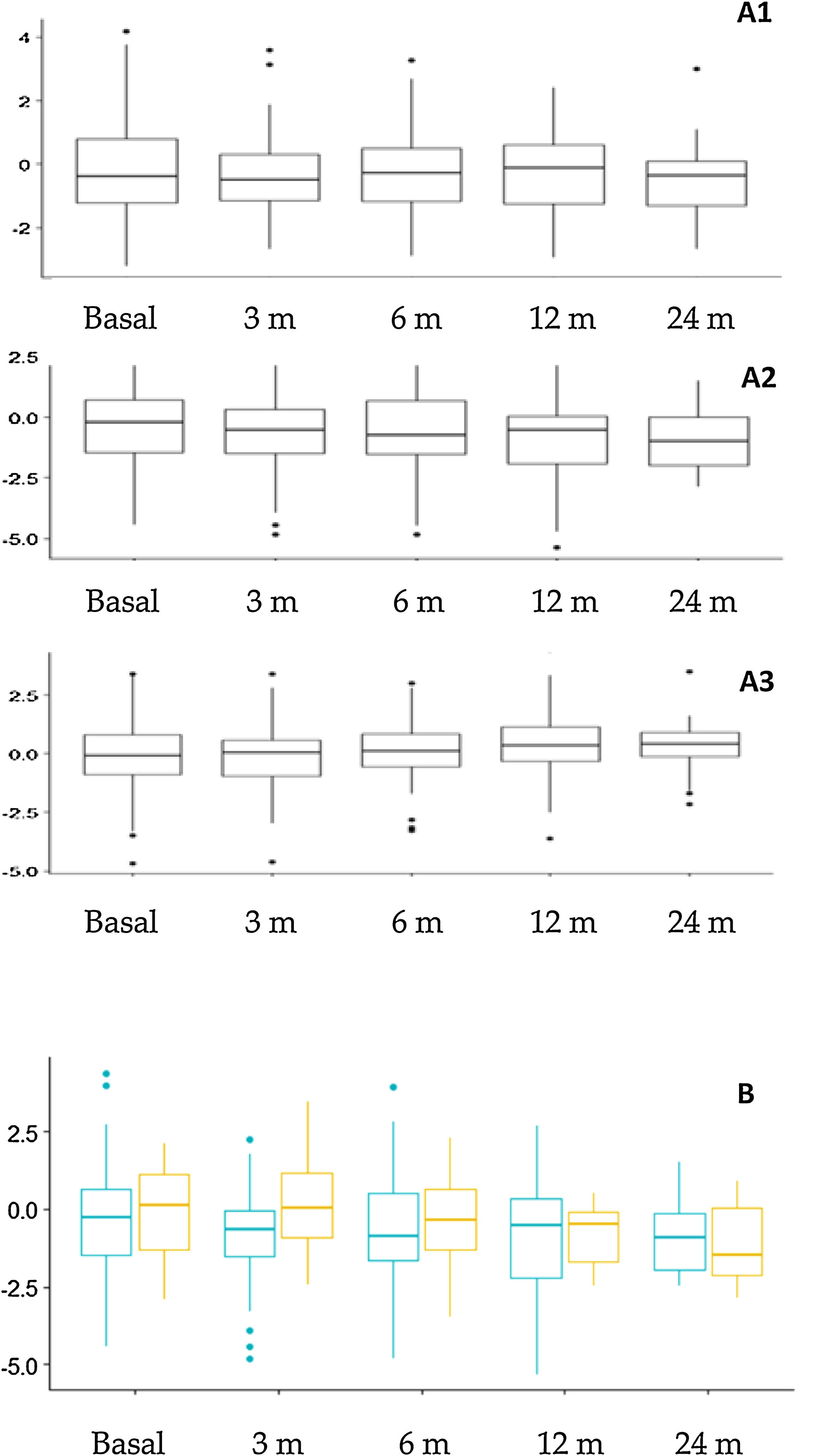
Cambios antropométricos. A. Evolución de la puntuación z de peso (A1), talla (A2) e índice de masa corporal (A3) a lo largo del seguimiento. B. Comparación de la evolución del z score de talla según la edad al inicio de TDC. Naranja (derecha): Pacientes que comenzaron TDC antes de los 2 años de edad. Azul (izquierda): pacientes que comenzaron TDC después de los 2 años de edad.
m: meses; TDC: terapias dietéticas cetogénicas.
Se observaron aumentos de vitamina D (p=0,05), ácido fólico (p<0,001), ferritina (p=0,009), selenio (p<0,001) y carnitina (p=0,008) a los 12 meses en comparación con el valor inicial, y de ferritina (p=0,047) y carnitina (p≤0,02) a los 24 meses. Se redujeron prealbúmina (p≤0,034), proteína transportadora de retinol (p≤0,025) y magnesio (p≤0,04) a los 12 y 24 meses y vitamina A (p=0,03) a los 12 meses.
DiscusiónEl presente estudio presenta la serie española más numerosa de pacientes pediátricos epilépticos tratados con TDC, y describe su eficacia y seguridad. En trabajos anteriores el 15% de los pacientes quedaban libres de crisis y>30% presentaba una reducción superior al 50%7,29–31. No se han encontrado diferencias de eficacia en función de la edad, el tipo de convulsiones o de la dieta4,7,8. En nuestra cohorte el 12-15% quedó libres de crisis y el 41-16% presentó buena respuesta. Observamos que la DCC fue más efectiva, pero solo a los 3 y 12 meses. Algunos estudios aleatorizados han mostrado esta tendencia, aunque sus resultados no fueron estadísticamente significativos8.
A pesar de ser un tratamiento eficaz, la ineficacia es el motivo más frecuente de abandono en nuestra serie. La selección del paciente es importante para el éxito de las TDC. Se han demostrado más beneficios en32: SD-GLUT1, deficiencia de piruvato deshidrogenasa (PDH), síndromes de Doose, Ohtahara, Dravet y Angelman, enfermedad mitocondrial por deficiencia del complejo I y esclerosis tuberosa. Una cuarta parte de nuestros niños presentaba alguna de estas entidades. Quince de los pacientes con esos trastornos recibieron la TDC durante al menos 2 años, y la reducción de las convulsiones fue superior al 90% en todos los casos. Los pacientes con enfermedad metabólica fueron los que con mayor frecuencia quedaron libres de crisis, probablemente porque la mitad de ellos padecían SD-GLUT1 o deficiencia de PDH. Las TDC son el gold estándar para ambos trastornos, en los que el metabolismo cerebral está comprometido y las TDC proporcionan un sustrato alternativo. Entre los pacientes con enfermedad estructural la dieta también fue eficaz, pero en menos de la mitad de los casos. En este grupo etiológico las TDC pueden servir de puente hasta poder realizar el tratamiento quirúrgico definitivo, permitiendo una mejoría, aunque sea parcial, de las crisis y, en ocasiones, una reducción de los FAE y sus EA.
Los lactantes también se benefician de las TDC. Observamos que el 16-28% quedaron libres de crisis, y el 24-60% tuvo una buena respuesta. Nordli et al.33 reportaron datos similares, y una revisión reciente concluye que las TDC, a pesar de lo que se pensaba en el pasado, son seguras y eficaces para el tratamiento de la epilepsia refractaria, el SD-GLUT1 y la deficiencia de PDH en niños≤2años34. En este grupo de edad debe considerarse su uso precozmente debido a que el cumplimiento es más fácil y la DCC debe ser de primera elección32.
Los EA no fueron una causa frecuente de retirada (excepto durante el primer mes) y fueron generalmente leves, transitorios y fáciles de tratar. Los EA digestivos fueron muy prevalentes, aunque menos que en otros estudios en los que ocurrieron en más de la mitad de los niños11,12. La hipercalciuria es un EA temido, ya que es asintomática y puede derivar en complicaciones graves como la nefrolitiasis. La hipercalciuria fue muy común, pero nuestra incidencia de litiasis fue mucho menor a lo reportado14. La monitorización permanente del cociente calcio/creatinina urinario y el uso temprano de citrato potásico cuando aparece la hipercalciuria probablemente sean las razones de nuestra baja incidencia. La dislipidemia fue también frecuente y se presentó precozmente, como en otras series15. El riesgo cardiovascular a largo plazo es controvertido, y se ha descrito la normalización del perfil lipídico a los 12 meses15. La hipercolesterolemia se produjo durante todo el seguimiento, mientras que nuestra incidencia de hipertrigliceridemia disminuyó al año. La dislipidemia pudo controlarse solo con modificaciones dietéticas, reemplazando las grasas saturadas (utilizadas para mejorar la palatabilidad de la dieta) por monoinsaturadas y poliinsaturadas.
Otra desventaja de las TDC es la restricción dietética. De hecho, la segunda razón para la retirada en nuestra cohorte fueron las dificultades de cumplimiento. Curiosamente, aunque la DCC es más restrictiva, el incumplimiento fue más frecuente entre los pacientes con DAM. La edad podría estar implicada en esta paradoja. La DCC suele indicarse en niños pequeños en los que es fácil controlar la alimentación, ya que se basa en fórmulas líquidas o alimentos triturados. A medida que el niño crece el cumplimiento puede volverse difícil. Esto, unido al hecho de que la DCC parece ser más eficaz en niños pequeños8, refuerza la indicación de DCC como primera opción en<2 años. Sin embargo, cuando aparecen dificultades de cumplimiento la sustitución de DCC o DAM por dietas menos restrictivas (como DBIG) puede ayudar a mantener el tratamiento.
El impacto de las TDC sobre el estado nutricional ha sido ampliamente estudiado16,17. La ingesta de calcio y vitamina D disminuye en las TDC. Además, sus niveles suelen estar bajos en niños con epilepsia grave, y esto puede conllevar la aparición de efectos graves como fracturas. En nuestra cohorte los niveles de selenio, carnitina y vitamina D, que generalmente se reducen con las TDC, mejoraron tras comenzar la dieta, ya que el déficit fue detectado basalmente y se prescribieron los suplementos pertinentes. Por ello, conviene destacar la importancia de una adecuada evaluación inicial. El efecto sobre el crecimiento es controvertido. El retraso del crecimiento puede afectar al 20-30%18–22,35, pero puede revertirse tras la retirada36. El tratamiento a largo plazo puede afectar significativamente a la talla, especialmente en los más pequeños37. Observamos diferencias estadísticamente significativas en los valores de la puntuación z de la talla durante el seguimiento, y el 15-24% de nuestros pacientes presentó valores <-2, especialmente los menores de 2 años al inicio de la TDC. Algunas causas relacionadas con el retraso del crecimiento incluyen la ingesta inadecuada de energía y proteínas, cetosis21,22, acidosis y cambios endocrinos35. No encontramos diferencias significativas en la evolución del crecimiento según el tipo de dieta, que fue suficiente en proteínas en todos los casos. Sin embargo, debe considerarse que los niños<2 años siguieron DCC, y es precisamente este grupo de pacientes el más vulnerable. Sería útil identificar a los sujetos con mayor riesgo de retraso de crecimiento antes del inicio de TDC (<2años) y durante el tratamiento (acidosis, cetosis, cambios endocrinos) para realizar un seguimiento más estrecho. Las TDC suelen mantenerse al menos 2 años. Sin embargo, en los espasmos infantiles (un tipo de convulsión altamente específica del periodo de lactante), no se encontraron diferencias en la tasa de recaída entre los pacientes libres de crisis que interrumpieron la dieta a los 8 meses o 2 años38. En vista del posible efecto sobre el crecimiento, especialmente en los lactantes, en estos casos se debe considerar una menor duración de la dieta39.
Los mecanismos de acción de las TDC continúan investigándose. La cetosis es uno de los más estudiados. Aunque menos de la mitad de nuestros pacientes presentaron niveles óptimos de beta-hidroxibutirato, la mayoría mejoraron. Esto sugiere la existencia de efectos anticonvulsivantes y neuroprotectores a través de otros mecanismos.
Nuestro trabajo presenta algunas limitaciones. Se trata de un estudio observacional y retrospectivo, el número de pacientes en cada grupo no fue el mismo y los protocolos de tratamiento utilizados no fueron exactamente iguales durante el período de estudio. Esto dificulta la extrapolación de los resultados a la práctica clínica, especialmente el hallazgo de que la DCC puede ser más eficaz. Además, es difícil determinar si las TDC son las únicas responsables del deterioro del crecimiento. La epilepsia en sí misma, enfermedades subyacentes y otros tratamientos pueden contribuir al impacto sobre el crecimiento.
En conclusión, las TDC son eficaces contra la epilepsia refractaria infantil. Los EA son frecuentes pero leves y fáciles de tratar. Los pacientes con TDC requieren un seguimiento estrecho por parte de personal cualificado con el fin de identificar y tratar los EA y controlar su crecimiento, ya que puede alterarse, especialmente en los niños de menor edad. Además, las TDC deben considerarse un tratamiento dinámico, pudiendo realizarse modificaciones dietéticas para reducir los EA, mejorar el cumplimiento o atenuar el impacto en el estado nutricional.
FinanciaciónEste estudio no ha sido financiado por ninguna entidad.
Conflicto de interesesLa Dra. Jana Ruiz Herrero ha participado en 2 cursos de dieta cetogénica patrocinados por Nutricia y ha recibido financiación para viajes de Nutricia.
La Dr. Cañedo Villarroya ha recibido honorarios y apoyo no financiero de Nutricia, Mead Johnson, Nestlé, Abbott y Orphan.
El Dr. García Peñas ha recibido honorarios de Nutricia, Sanofi, UCB, Eisai/Esteve, Bial y GW.
La Sra. Puerta Macfarland ha participado en 2 cursos de dieta cetogénica patrocinados por Nutricia.
La Dra. Consuelo Pedrón Giner ha desempeñado labores de consultora y ha recibido honorarios por conferencias de Nutricia, Vitaflo, Nestlé y Mead Jhonson; y ha recibido financiación para viajes de Nutricia, Vitaflo y Nestlé.
Las Sra. García Alcolea y Gómez Fernández no tienen conflicto de intereses que declarar.
