



Langerhans cell histiocytosis (LCH) is a type of myeloid neoplasia that can affect different organs or tissues and exhibits substantial variability in its clinical presentation and biological behaviour, so it may mimic different diseases. Performance of different clinical assessments and laboratory and imaging tests is recommended to determine the extent of involvement, which may be of a single location or multisystemic, and the presence or absence of dysfunction in risk organs, such as the haematopoietic system, liver and spleen. The diagnosis must be confirmed by histological examination of a biopsy sample. Molecular tests have identified mutations in the mitogen-activated protein kinase (MAPK) pathway, which has expanded treatment options. The diagnosis is complex and there is controversy regarding the management of certain cases. Treatment recommendations depend on the location of the lesions and the extent of involvement. International collaborative studies have demonstrated the effectiveness of prolonged combination therapies such as vinblastine and prednisone in severe or multisystemic forms, and anti-inflammatory drugs such as indomethacin and other cytostatic combinations have proven beneficial. Langerhans cell histiocytosis is a good example of the importance of precision medicine and the benefit of identifying molecular targets, common to different neoplasms, to develop new therapies. MAPK pathway inhibitors offer an alternative treatment option in refractory cases and neurodegenerative forms of LCH. Molecular testing can contribute to the prognosis, treatment and follow-up of LCH, especially in severe forms of disease.
La histiocitosis de células de Langerhans es un tipo de neoplasia hematológica de origen mieloide, que puede afectar a diferentes órganos o tejidos, con gran variabilidad en la presentación clínica y comportamiento biológico, por lo que puede simular diferentes enfermedades. Se recomienda realizar diversas pruebas clínicas, analíticas y de imagen, para determinar la extensión de la afectación, que puede ser única o multisistémica, y la presencia o no de disfunción en órganos de riesgo como sistema hematopoyético, hígado y bazo. El diagnóstico se debe confirmar mediante biopsia y estudio histológico. Los estudios moleculares han permitido identificar mutaciones en la vía MAPK, lo que han ampliado las opciones terapéuticas. El diagnóstico es complejo y existe controversia en el manejo de ciertos casos. Las recomendaciones terapéuticas dependen de la localización de las lesiones y la extensión de la afectación. Los estudios colaborativos internacionales han demostrado la efectividad de terapias prolongadas combinadas como vinblastina y prednisona en formas graves o multisistémicas y destaca el papel beneficioso de fármacos antinflamatorios como indometacina y de otras combinaciones de citostáticos. HCL representa un buen ejemplo de la importancia de la medicina de precisión y del beneficio de la identificación de dianas moleculares, comunes a diferentes neoplasias, para desarrollar nuevas terapias dirigidas. Los inhibidores de la vía MAPK representan una alternativa terapéutica en casos refractarios y en las formas neurodegenerativas de HCL. Los estudios moleculares pueden contribuir en el pronóstico, tratamiento y seguimiento, especialmente en las formas graves.
The histiocytoses are a heterogeneous group of diseases that pose a diagnostic challenge and the management of which continues to be subject to debate.1–3 In the past 10 years, advances in the knowledge of their pathogenesis, the cells of origin and the molecular pathways involved have led to changes in their classification and the recommendations for diagnosis and treatment.1–3 In the paediatric age group, the most important may be Langerhans cell histiocytosis (LCH), which is usually associated with genetic changes in the mitogen-activated protein kinase (MAPK) pathway.3–6 The dysregulation of this molecular pathway is a key mechanism in the oncogenesis of different cancers.7 The treatment of LCH is based on anti-inflammatory agents, such as steroids, and cytostatic agents, such as vinblastine, but recently new therapeutic strategies have been developed, such as MAPK inhibitors, for severe and refractory forms of disease.7–11 Langerhans cell histiocytosis is a good example of the importance of precision medicine and the benefits of identifying molecular targets common to different malignancies in order to develop new targeted therapies.10,11 Molecular testing is important to establish the prognosis and treatment of affected patients.
PathophysiologyThe broad spectrum of manifestations and heterogeneity of the clinical course of LCH, ranging from self-healing lesions to life-threatening multisystemic disease, have kept this disease in obscurity and contributed to its original labelling as histiocytosis X.2 There are 2 main broad theories about its pathogenesis: one, that it is a reactive process due to inappropriate immune stimulation, and the other, that it is a neoplastic disorder.
The immune dysregulation hypothesis is based on the benign morphology of the proliferating cells, the associated inflammatory infiltrate, cellular heterogeneity, low proportion of abnormal histiocytes in lesions, production of local and systemic cytokines and the tendency toward spontaneous resolution. In this inflammatory environment, T cells and cytokines play key roles that contribute to the clinical manifestations and the development of lesions with fibrosis, necrosis and osteolysis.12,13
The identification of genetic changes supports the current clonal and neoplastic hypothesis. In 2010, Badalian-Very et al. described abnormal CD1a+ CD207+ histiocytes that carried a somatic variant of the BRAFV600E oncogene in 57% of 61 affected patients.14 This variant has been confirmed in numerous cohorts, such as a paediatric case series in France that included 315 patients, with detection of BRAFV600E in 54.6%.15 Other changes in the BRAF gene have been described with lesser frequency. Molecular findings have guided the new classification of histiocytic disorders.3 At present, according to the World Health Organization, LCH is considered a myeloid neoplasia.16
There has also been substantial controversy regarding the cell of origin. In patients with severe forms, genetic changes have been found in haematopoietic stem cells in the bone marrow and yolk sack, while in less severe forms, they have been detected in the affected tissue. These findings suggest that genetic changes may take place at different points in development and in different types of cells.4,17
Other activating pathogenic variants have been found in the RAS-RAF-MEK-ERK pathway involved in the proliferation, survival, differentiation and activation of myeloid dendritic cell precursors. Brown et al. found changes in the MAP2K1 gene in 27.5% of cases of LCH in the absence of BRAF variants.18 Mutations in other genes, such as ALK, ARAF, ERBB3, PIK3KCA and MAP3K1, have also been reported. Variants are not detected in approximately 10%–20% of patients, which has led to the formulation of additional aetiological hypotheses, such as copy number variations or epigenetic changes.4–6
The clinical heterogeneity and the different forms of LCH are not only explained by the genetic changes found in the MAPK pathway, but also by the cell of origin where these variants arise. Thus, high-risk disease forms have been described in patients that had these variants in myeloid progenitor cells, such as CD11c+ dendritic cells or CD14+ monocytes and CD34+ haematopoietic stem cells in the bone marrow. In contrast, in less severe forms of LCH, these changes have been described in cells residing in the tissues of each affected organ. Thus, the LCH pathogenesis model is an integrated model that considers the abnormal activation of ERK and signalling pathways for the proliferation, survival, differentiation and activation of myeloid dendritic cell precursors (Fig. 1). It has also been hypothesised that changes in this pathway may alter the cytokine response induced by toll-like receptors (TLRs) in dendritic cells.2,4–6,12,13
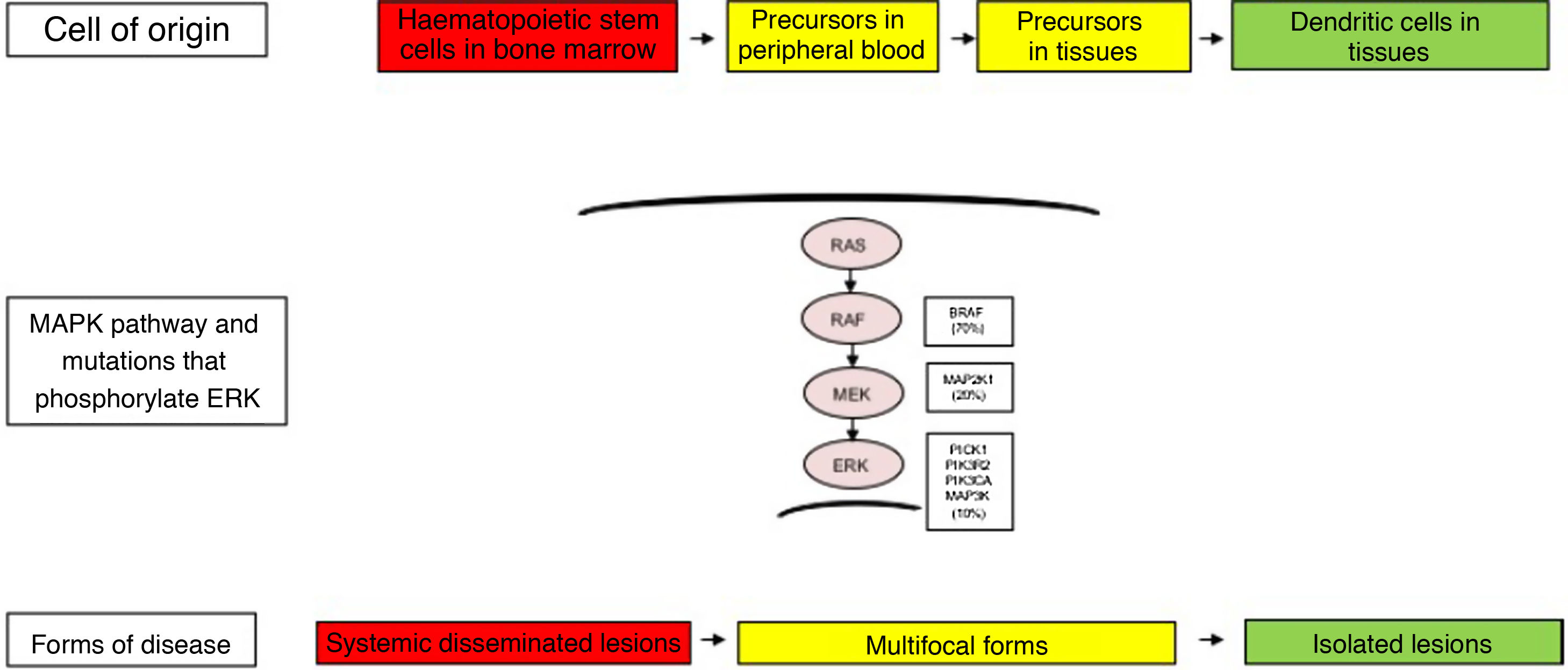
Cellular and molecular model of the development of the different forms of Langerhans cell histiocytosis. The constitutive activation of the ERK pathway due to different genetic changes in haematopoietic stem cells or myeloid precursors causes multisystemic disseminated disease, while changes in precursor cells in the peripheral blood or tissue causes multifocal disease and changes in more differentiated cells in specific tissues lead to single lesions. MAPK, mitogen-activated protein kinase.
Langerhans cell histiocytosis is considered one of the greatest mimics, as it may be confused with many other diseases. The differential diagnosis is broad, complex and requires exclusion of other malignant or infectious diseases. Given its low incidence, it is important to maintain a high level of suspicion.19
The history-taking and physical examination must explore signs and symptoms deriving from skin lesions, bone and muscle pain, fever, lymph node enlargement, cytopenias, respiratory symptoms, persistent diarrhoea, abdominal organ enlargement, polyuria/polydipsia, faltering growth, neurologic abnormalities etc. Any organ may be affected, there can be one or multiple lesions, and the lesions may involve a single system or multiple ones (multisystemic). Skin and bone lesions are most frequent.4–6,20–22
The definitive diagnosis requires a biopsy of the most accessible lesion for confirmation through histological examination and immunohistochemistry. Pathological Langerhans cells express S-100 protein, CD1a and CD207 (langerin), and are associated with inflammatory granulomatous infiltrates of macrophages, lymphocytes and eosinophils. The most specific markers of LCH are CD1a and CD207, but they are also expressed by other mononuclear precursor cells. Therefore, diagnosis requires interpretation of the histological findings in the context of the clinical and radiological findings.3–6
The histological examination is indispensable to make the diagnosis. Vertebral involvement (“vertebra plana” or lesions in the upper portion of the cervical spine without involvement of soft tissues) with a high surgical risk is one of the exceptions warranting performance of a biopsy before initiating treatment, as long as other possible causes are ruled out, especially malignant and infectious aetiologies.20–23
At present, molecular tests are recommended to identify changes in the BRAF gene (BRAFV600E) and other genes such as MAP2K1.3,14,18 These tests can support the diagnosis and are relevant to the prognosis and management, and may expand treatment options to include targeted therapies, especially in patients with organic dysfunction and risk organ involvement (liver, spleen, blood) or in refractory cases.4,10,11,24 A cohort study in France found that patients with the BRAFV600E variant exhibited more severe disease and a poorer response to first-line treatment.15 Other studies have found a correlation between the presence of BRAFV600E in blood and a higher rate of recurrence.25,26
After diagnosis, it is important to assess the extent of involvement to determine the severity of disease and whether it affects multiple systems. Involvement of risk organs (haematopoietic system, liver, spleen) must be ruled out, which usually manifests with cytopaenia (in at least 2 lineages: haemoglobin <10 g/dL, leukocyte count <4000 cells/mm3, platelet count <100 000/mm3), hepatomegaly and/or splenomegaly (defined as organ edge 3 cm or 2 cm, respectively, below the costal margin) or liver dysfunction (protein <5.5 g/dL, albumin <2.5 g/dL, elevation of gamma-glutamyl transpeptidase [GGT]).4–6,9,20–22
Performance of laboratory tests is recommended, including a complete blood count, coagulation tests, electrolyte panel, erythrocyte sedimentation rate, liver and renal function tests, protein electrophoresis and iron panel, although the results are nonspecific.21 The imaging tests should include an abdominal ultrasound and a plain X-ray, computed tomography (CT) scan or magnetic resonance imaging (MRI) scan of suggestive bone lesions and other bones.20–22 There is controversy regarding the gold standard for assessment of lytic lesions, as in general they can be easily detected by plain radiography, but they may be difficult to visualize in locations like the ribs, spine or pelvis. Computed tomography or MRI scans are useful for more thorough assessment of bone and soft tissue lesions. The role of 18-fluorodeoxyglucose (18F-FDG) positron-emission tomography (PET) CT is not well established, but this is a very sensitive method that could be useful in complex or recurrent cases.21,27 Other studies should be performed based on the symptoms and the suspected affected organs to establish involvement: endocrine/metabolic assessment tests (e.g. water deprivation test), craniospinal or hypothalamic-pituitary MRI, hearing test, sensory evoked potentials, neurocognitive and psychometric evaluation, pulmonary CT, lung function tests, bronchoalveolar lavage, gastroscopy, colonoscopy, etc.4–6,20–22
During the follow-up, different tests should be conducted depending on the location of the lesions. It must be taken into account that the response bone lesions may take months to be evident in imaging tests, and it is recommended that the same methods are used for assessment throughout the follow-up. The radiological follow-up is usually limited to the assessment of the anatomical regions affected at diagnosis, unless there are clinical manifestations suggesting the presence of new lesions.22,23
Management of mild forms: cutaneous involvement, isolated bone lesions, bone lesions in special locations and isolated involvement in other sitesIn general, patients with disease involving a single system and confined to a single location only require local treatment or observation after the initial biopsy. Single lesions have a good prognosis and may resolve spontaneously.20–23
Isolated cutaneous involvementSkin lesions are frequent and varied, although exclusive cutaneous involvement is rare (5%). They may appear at any age, but characteristically develop in newborns and infants. They tend to manifest as seborrheic eczema or persistent cradle cap, lesions in skinfolds and the perineum or generalised rash. They may have papules, pustules, vesicles or ulcers with bleeding scabs. The recommended management of LCH with exclusive cutaneous involvement is observation and close monitoring, as it may resolve without treatment but there is also a risk of reactivation or progression to multisystemic disease in up to 40% of cases, especially in carriers of the BRAFV600E variant. In the case of single lesions, surgical resection for the purpose of diagnosis is usually curative.28–31
Pharmacological treatment is only recommended for symptomatic patients, but there are no set care standards for forms with isolated cutaneous involvement. The main treatment options are20–24,32,33:
- •
Topical steroid therapy with agents of medium to high potency is the first-line treatment, but may not always be effective, and recurrence is common after discontinuation. Short courses of oral steroid therapy may also be given.
- •
Methotrexate, mercaptopurine or oral hydroxyurea. Possible administration of methotrexate at 20 mg/m2/week, mercaptopurine at 50 mg/m2/day or hydroxyurea at 20 mg/kg/day, for 6–12 months, alone or in combination in refractory cases. Oral thalidomide (dose, 50−200 mg/day) may also be effective in children and adults.
- •
Other treatments in refractory cases: topical administration of nitrogen mustard or imiquimod or psoralene plus ultraviolet B phototherapy (PUVB) may be effective and safe in treatment-resistant forms when delivered in specialised facilities. Symptomatic cases with extensive cutaneous involvement may be treated with systemic chemotherapy (combination therapy with steroids and vinblastine for 6–12 months).
Bone lesions are present in approximately 80% of cases and may develop in any bone of the body, although they are rare in the hands and feet. The most frequently involved site is the skull, followed by the spine, extremities and pelvis. Bone lesions may be asymptomatic or be associated with pain, swelling or soft tissue or impaired mobility. They are usually lytic lesions with soft tissue mass.19–23
Single bone lesions (50%) can regress spontaneously, and observation and follow-up may be sufficient. Many lesions improve after the biopsy. Decisions regarding treatment should be based on the clinical manifestations, size and location of the lesion, with particular consideration of the potential compression of the spine, the presence of unacceptable deformities, severe pain and functional impairment. The possible treatments depend on the type of lesion19–23,30–37:
- -
Isolated cranial lesions in the calvarium or other cranial bones:
- •
Initial conservative management. Watchful waiting for spontaneous regression after biopsy.
- •
Curettage/surgery. Complete resection of small bone lesions (<2 cm) may be indicated when performing the biopsy. However, radical resection of large lesions (> 5 cm) is not indicated because it enlarges the bone defect, prolongs healing times and could cause permanent skeletal defects. For lesions measuring 2–5 cm in diameter, partial biopsy or curettage are sufficient. Intralesional injection of steroids during surgery has been found to have a prolonged pharmacological effect, alleviating pain and improving mobility with good tolerance in patients with easily accessed symptomatic lesions.
- •
Oral indomethacin. It has proven effective as first-line treatment, for pain management in recurrent disease and for disease reversion, achieving a response in more than 90% of symptomatic bone lesions with minimal adverse effects. The recommended dose is 1–2 mg/kg/day for a median of 6–12 months.35,36
- -
Skull lesions involving the facial skeleton, orbital socket, mastoid process and temporal bone. Involvement of facial bones and the anterior and middle cranial fossae (temporal bone, orbital socket, sphenoid, ethmoid and zygomatic bones) with intracranial tumour formation constitutes a risk to the central nervous system (CNS) and may be associated with a higher probability of diabetes insipidus and neurodegenerative disease.36 Systemic chemotherapy with vinblastine and prednisone (stratum I of the current LCH-IV protocol) is generally recommended, but there is some controversy surrounding this approach, as good outcomes may be achieved in some lesions with watchful waiting or indomethacine.32,35,36
- -
Vertebral lesions. Any vertebra may be involved, although cervical involvement is most frequent, with flattening of the vertebral body (vertebra plana). These lesions are usually painful and in rare cases be associated with neurologic impairment due to extension of the mass into the soft tissues and spinal compression. The current recommendation is watchful waiting except in the case of extension of the mass to the soft tissue or the intraspinal space, which may require chemotherapy. Orthopaedic treatment with support corsets may be necessary in case of vertebral instability or neurologic manifestations.
The risk of reactivation of a single bone lesion is low (10%). In the case of recurrent, refractory or progressing lesion, the treatment is based on the administration of indomethacin or chemotherapy (prednisone + vinblastine, cytarabine + vincristine, cladribine, hydroxyurea). There are also cases of recurrent mild isolated bone lesions that regress spontaneously, and bisphosphonates may be effective in some cases.20–23
Lymph node involvementSurgical excision is the standard of care in the case of isolated lymph node involvement.32
Isolated pulmonary involvementIn adolescents, smoking cessation is key to achieve a cure. Chemotherapy is recommended in symptomatic cases.
Space-occupying lesions in the central nervous systemThey may develop anywhere, but they are most frequent in the hypothalamic-pituitary region, with pituitary stalk thickening. In some locations, surgical resection may be diagnostic as well as curative. Other lesions tend to respond to chemotherapy (vinblastine/steroids, vincristine/cytarabine, cladribine as monotherapy).39
Central diabetes insipidus (CDI) may precede or follow the development of LCH. Affected patients are at increased risk of neurodegenerative disease, but specific treatments for LCH cannot revert CDI, so their use is not recommended.32,38
Management of multisystemic forms and organ dysfunctionPatients are classified into risk groups based on disease extension and the affected organs. Involvement of multiple systems indicates the need to eliminate the clonal precursors with systemic chemotherapy. Treatment with steroids and vinblastine has been found to achieve excellent responses in patients with multisystemic disease and is recommended as first-line treatment by the Histiocyte Society.8,9,31
Due to the low prevalence of LCH, multicentre studies have been key to assess outcomes and establish treatment recommendations. In Spain, we ought to highlight the work of the Working Group on Histiocytosis of the Sociedad Española de Hematología y Oncología Pediátricas (SEHOP, Spanish Society of Paediatric Haematology and Oncology) and the participation in collaborative studies at the national (LCH-95, LCH-2000) and international level (LCH-III, LCH-IV). The published results of the LCH-III trial in 554 paediatric patients with multisystem LCH (MS-LCH) show an overall survival of 99% in patients without risk organ involvement and of 84% in patients with risk organ involvement, with an improvement in the prognosis of MS-LCH compared to previous trials (LCH-I, LCH-II). The addition of methotrexate was not beneficial. It was found that prolonging treatment was beneficial for prevention of recurrence.9 A recent study has found a negative impact on patient outcomes of gastrointestinal involvement in MS-LCH.37
The current International Collaborative Treatment Protocol trial (LCH-IV, ClinicalTrials.gov: NCT02205762) includes recommendations for all forms of LCH, including cases of first-line treatment failure and reactivation. The SEHOP oversees the trial in Spain, with participation of 27 hospitals coordinated from the Instituto Investigación Sanitaria BioCruces Bizkaia, with the support of the ECLIM-SEHOP and SCReN platforms. Since 2016, this trial has included 103 patients managed in 23 hospitals in Spain.
Despite good overall survival outcomes, more than 40% of patients with MS-LCH are refractory to first-line treatment or experience recurrence, and therefore require other treatments. Different salvage treatments have been used traditionally, such as cytarabine, cladribine, clofarabine and haematopoietic stem cell transplantation. Once genetic changes were detected in the MAPK/ERK pathway, the therapeutic armamentarium has broadened to include approaches such as targeted therapy and MAPK/ERK pathway inhibitors, which have achieved high response rates in children with refractory high-risk LCH.4–7,39
Several studies have described treatment with vemurafenib and other inhibitors (dabrafenib, cobimetinib, trametinib), highlighting the excellent response rates and the quick effect in severe forms of disease.7,10,11,32,39 However, this is not a curative treatment, as relapse nearly always succeeds discontinuation. Clinical trials are required to confirm their efficacy and safety, the optimal timing and duration for their use and the usefulness of combination therapy with MAPK/ERK pathway inhibitors and chemotherapy.
Followup: long-term sequelae and neurodegenerationReactivation, involvement of high-risk organs, long-term sequelae and neurodegeneration associated with LCH pose the greatest challenges in diagnosis and treatment. Approximately 50% of patients with LCH develop some form of sequelae, especially in the case of MS-LCH. Reactivations and high-risk CNS lesions are associated with a higher incidence of long-term complications. Sequelae develop most frequently in skin, bone and the hypothalamic-pituitary region, although they may affect other sites.22,38
Orthopaedic sequelae (secondary to pathologic long bone fractures, deformities, scoliosis) are frequent in the case of bone lesions (20%–30% of patients, especially in children aged less than 3 years). Cranial bone involvement may cause proptosis, facial asymmetry, hearing loss, tooth losses, etc.22,35,38 Skin lesions may result in ulcerations, scarring and other cosmetic sequelae.28,29
Involvement of the hypothalamic-pituitary axis may produce permanent endocrine sequelae, such as diabetes insipidus, that do not respond to treatment of LCH. Anterior pituitary hormone deficiencies are less frequent, but can cause growth delay, hypothyroidism, hypogonadism, amenorrhoea and early or delayed puberty.38
Neurodegenerative central nervous system involvement (ND-LCH) is a progressive syndrome characterised by radiological abnormalities and clinical features with little correlation between the two. It is a late-onset and progressive form of disease characterised by involvement of the cerebellum and brainstem that may manifest with neurocognitive impairment, which usually develops between 5 and 15 years following the initial diagnosis. The incidence ranges from 1% to 24%, depending on known risk factors such involvement of the facial skeleton, presence of CDI and the BRAFV600E variant, all of which increase its probability. The diagnosis of ND-LCH is complex and treatment recommendations are not yet well established. Different treatment strategies with immunomodulating and cytostatic agents have been proposed, but achieved poor responses, and MAPK inhibitors may be used in select cases.40
FundingThe study was partially funded by a grant allocated to the Histiocytosis Research Project of the Fundación Vasca de Innovación e Investigación Sanitaria BIOEF (BIO16/ER/020/BC) and the Asociación Española contra la Histiocitosis-ACHE (BC/A/15/012, BIOEF11/017, BIOEF09/047), whose principal investigator is Itziar Astigarraga.
Conflicts of interestThe authors have no conflicts of interest to declare.
We thank the patients, family members and professionals that have participated in the collaborative studies on histiocytosis. We also thank Ana Barriuso for her technical help in developing the manuscript, Marta Llarena, Ane Karmele Sáenz de Arzamendi and the Spanish Clinical Research Network (SCReN) platform of the Instituto de Salud Carlos III for their support and monitoring of the international LCH-IV study. We thank the ECLIM-SEHOP for the help in the implementation and development of international multicentre childhood cancer clinical trials in Spain.
Please cite this article as: Astigarraga I, García-Obregón S, Pérez-Martínez A, Gutiérrez-Carrasco I, Santa-María V, Rodríguez-Vigil Iturrate C, et al. Histiocitosis de células de Langerhans. Avances en la patogenia y práctica clínica. An Pediatr (Barc). 2022;97:130.
