



La histiocitosis de células de Langerhans (HCL) es un tipo de neoplasia hematológica de origen mieloide, que puede afectar a diferentes órganos o tejidos, con gran variabilidad en la presentación clínica y comportamiento biológico, por lo que puede simular diferentes enfermedades. Se recomienda realizar diversas pruebas clínicas, analíticas y de imagen, para determinar la extensión de la afectación, que puede ser única o multisistémica, y la presencia o no de disfunción en órganos de riesgo como sistema hematopoyético, hígado y bazo. El diagnóstico se debe confirmar mediante biopsia y estudio histológico. Los estudios moleculares han permitido identificar mutaciones en la vía MAPK, lo que han ampliado las opciones terapéuticas. El diagnóstico es complejo y existe controversia en el manejo de ciertos casos. Las recomendaciones terapéuticas dependen de la localización de las lesiones y de la extensión de la afectación. Los estudios colaborativos internacionales han demostrado la efectividad de terapias prolongadas combinadas como vinblastina y prednisona en formas graves o multisistémicas y destaca el papel beneficioso de fármacos antiinflamatorios como indometacina y de otras combinaciones de citostáticos. La HCL representa un buen ejemplo de la importancia de la medicina de precisión y del beneficio de la identificación de dianas moleculares, comunes a diferentes neoplasias, para desarrollar nuevas terapias dirigidas. Los inhibidores de la vía MAPK representan una alternativa terapéutica en casos refractarios y en las formas neurodegenerativas de la HCL. Los estudios moleculares pueden contribuir en el pronóstico, el tratamiento y el seguimiento, especialmente en las formas graves.
Langerhans cell histiocytosis (LCH) is a type of myeloid neoplasia that can affect different organs or tissues and exhibits substantial variability in its clinical presentation and biological behaviour, so it may mimic different diseases. Performance of different clinical assessments and laboratory and imaging tests is recommended to determine the extent of involvement, which may be of a single location or multisystemic, and the presence or absence of dysfunction in risk organs, such as the haematopoietic system, liver and spleen. The diagnosis must be confirmed by histological examination of a biopsy sample. Molecular tests have identified mutations in the mitogen-activated protein kinase (MAPK) pathway, which has expanded treatment options. The diagnosis is complex and there is controversy regarding the management of certain cases. Treatment recommendations depend on the location of the lesions and the extent of involvement. International collaborative studies have demonstrated the effectiveness of prolonged combination therapies such as vinblastine and prednisone in severe or multisystemic forms, and anti-inflammatory drugs such as indomethacin and other cytostatic combinations have proven beneficial. LCH is a good example of the importance of precision medicine and the benefit of identifying molecular targets, common to different neoplasms, to develop new therapies. MAPK pathway inhibitors offer an alternative treatment option in refractory cases and neurodegenerative forms of LCH. Molecular testing can contribute to the prognosis, treatment and follow-up of LCH, especially in severe forms of disease.
Las histiocitosis son un grupo heterogéneo de enfermedades que representan un reto diagnóstico y plantean controversias en el manejo clínico1–3. En los últimos 10 años, los avances en el conocimiento patogénico, identificación de células de origen y vías moleculares, han generado cambios en la clasificación, recomendaciones diagnósticas y terapéuticas1–3. En la edad pediátrica destaca la histiocitosis de células de Langerhans (HCL) que suele asociar mutaciones en la vía de señalización dependiente de la proteína quinasa de activación mitogénica (MAPK)3–6. La desregulación de esta vía molecular es un mecanismo fundamental en la oncogénesis de diferentes neoplasias7. La terapia para la HCL se basa en fármacos antiinflamatorios, como corticoides, y citostáticos como vinblastina, pero recientemente se han desarrollado nuevas estrategias terapéuticas, como loa inhibidores MAPK, para formas graves y refractarias7–11. La HCL representa un buen ejemplo de la importancia de la medicina de precisión y del beneficio de la identificación de dianas moleculares, comunes a diferentes neoplasias, para desarrollar nuevas terapias dirigidas10,11. Los estudios moleculares son relevantes para el pronóstico y el tratamiento de los pacientes.
Bases patogénicasLa gran heterogeneidad clínica y variabilidad en el comportamiento biológico de la HCL, desde lesiones con resolución espontánea hasta trastornos multisistémicos amenazantes para la vida, han mantenido el enigma sobre esta enfermedad y apoyado su denominación inicial como histiocitosis X2. Destacan 2 grandes teorías patogénicas: reactiva por estimulación inmune inapropiada o neoplásica.
La teoría de desregulación inmune se apoyaba en la morfología benigna de las células que proliferan, infiltrado inflamatorio asociado, heterogeneidad celular, baja proporción de histiocitos patológicos en lesiones, producción de citoquinas (locales y sistémicas) y tendencia a resolución espontánea. En este microambiente inflamatorio destaca el papel de linfocitos T y citoquinas que contribuyen a la clínica y al desarrollo de lesiones con fibrosis, necrosis y osteolisis12,13.
La identificación de mutaciones apoya la consideración actual clonal y oncológica. En 2010, Badalian-Very et al. describieron histiocitos patológicos CD1a+ CD207+ portadores de mutación somática del oncogén BRAFV600E, en el 57% de 61 pacientes afectos14. Esta mutación genética ha sido confirmada en numerosas cohortes, como la serie pediátrica francesa de 315 pacientes que detecta BRAFV600E en el 54,6%15. También se han descrito otras alteraciones de BRAF con menor frecuencia. Los hallazgos moleculares han orientado la nueva clasificación de las histiocitosis3. Actualmente, según la OMS, la HCL se considera una neoplasia hematológica de origen mieloide16.
También ha habido mucha controversia sobre la célula de origen. En pacientes con formas clínicas graves se han detectado mutaciones en progenitores hematopoyéticos de médula ósea y saco vitelino, mientras que en formas menos graves se han descrito circunscritas al tejido afectado. Estos hallazgos sugieren que las mutaciones pueden ocurrir en diferentes momentos evolutivos y tipos celulares4,17.
Se han descubierto otras alteraciones patogénicas que condicionan la activación de la vía RAS-RAF-MEK-ERK que genera señales de proliferación, supervivencia, diferenciación y activación de precursores de células dendríticas mieloides. Brown et al., demostraron mutaciones en MAP2K1 en el 27,5% de casos de HCL, en ausencia de mutaciones de BRAF18. Además, se han descrito mutaciones en otros genes como ALK, ARAF, ERBB3, PIK3KCA y MAP3K1. En alrededor del 10-20% de los pacientes no se detecta ninguna mutación y se postulan mecanismos adicionales como cambios en el número de copias o alteraciones epigenéticas4–6.
Las diferentes formas clínicas heterogéneas de la HCL, no solo se explican por las mutaciones encontradas en la vía MAPK, sino también por el tipo celular donde surgen estas alteraciones genéticas. Así, se ha descrito cómo los pacientes con formas clínicas de alto riesgo presentan estas mutaciones en precursores mieloides, como células dendríticas (CD)11c+ o monocitos CD14+, y en médula ósea en progenitores hematopoyéticos CD34+. Mientras que en las formas menos graves de HCL estas alteraciones se han descrito en células residentes de cada órgano afecto. Por tanto, el modelo de patogénesis de la HCL es un modelo integrador que considera la activación patológica de ERK que genera señales de proliferación, supervivencia, diferenciación y la activación de los precursores de CD mieloides (fig. 1). También se plantea que las mutaciones en esta vía pueden alterar la respuesta de citoquinas inducida por los receptores tipo toll (TLR) en las células dendríticas2,4–6,12,13.
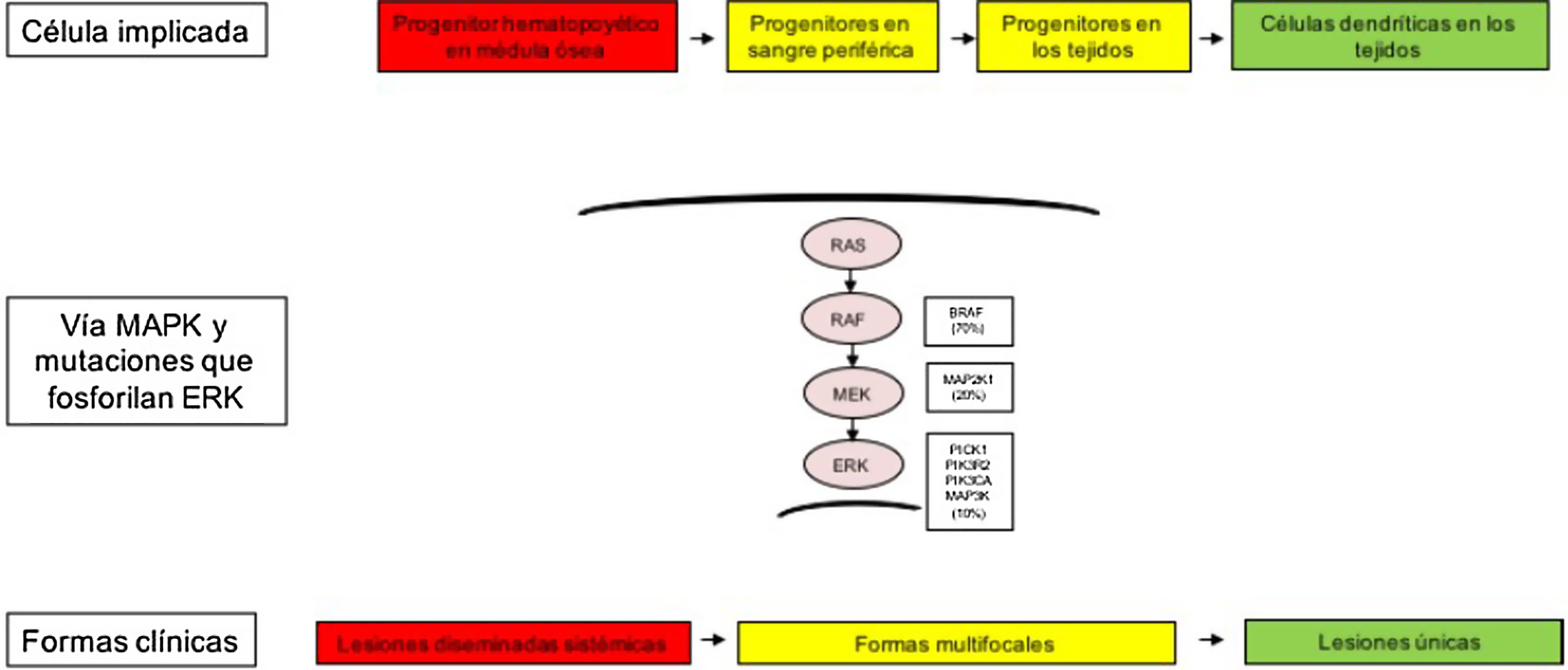
Modelo celular y molecular de desarrollo de las diferentes formas clínicas de la histiocitosis de células de Langerhans. La activación de la vía ERK por diferentes mutaciones en los precursores hematopoyéticos o precursores mieloides origina enfermedad diseminada multifocal, mientras que las mutaciones en las células precursoras en sangre periférica y tejidos origina enfermedad multifocal, mientras que las mutaciones en las células más diferenciadas de los tejidos lesiones únicas. MAPK: del inglés Mitogen-activated protein kinases.
La HCL se considera una de las enfermedades más «simuladoras», puede confundirse con otras muchas enfermedades. El diagnóstico diferencial es amplio, dificultoso y se deben descartar especialmente otros procesos malignos o infecciosos. Dada su escasa incidencia, es importante su sospecha clínica19.
La anamnesis y exploración física deben recoger síntomas y signos derivados de lesiones en piel, dolores osteomusculares, fiebre, adenomegalias, citopenias, síntomas respiratorios, diarrea persistente, megalias abdominales, poliuria/polidipsia, fallo de medro, cambios neurológicos, etc. Cualquier órgano puede estar afectado y las lesiones pueden afectar a un único sistema o varios (multisistémico), ser aisladas o múltiples. Las lesiones cutáneas y óseas son las más frecuentes4–6,20–22.
El diagnóstico definitivo exige biopsia de la lesión más accesible para la confirmación histológica e inmunohistoquímica. Las células de Langerhans patológicas expresan proteína S-100, CD1a y CD207 (langerina), y se acompañan de un infiltrado granulomatoso inflamatorio de macrófagos, linfocitos y eosinófilos. Los marcadores más específicos de HCL son CD1a y CD207, pero también son expresados por diferentes precursores mononucleares. Por ello, el diagnóstico histológico debe correlacionarse con los hallazgos clínicos y radiológicos3–6.
El estudio histológico es imprescindible para establecer el diagnóstico. La afectación vertebral (vértebra en galleta/plana o lesión cervical alta sin afectación de partes blandas) con alto riesgo quirúrgico representa una de las excepciones para realizar biopsia previa a iniciar tratamiento, siempre que se descarten otras causas, especialmente malignas o infecciosas20–23.
Actualmente se recomienda realizar estudios moleculares, para identificar mutaciones en BRAFV600E y otros genes como MAP2K13,14,18. Estos estudios moleculares apoyarían el diagnóstico y tienen relevancia clínica y pronóstica, así como amplían la posibilidad de tratamientos dirigidos sobre todo para pacientes con disfunción orgánica y afectación de órganos de riesgo (hígado, bazo y sangre) o en situaciones de refractariedad4,10,11,24. En una cohorte francesa se observó que pacientes con mutación BRAFV600E presentaban una enfermedad más grave y con peor respuesta al tratamiento de primera línea15. Otros estudios han mostrado la correlación entre positividad de BRAF en sangre y mayor tasa de recaídas25,26.
Tras el diagnóstico, es importante determinar la extensión de la afectación para valorar si la enfermedad es multisistémica y establecer la gravedad. Se debe descartar la afectación de órganos de riesgo (sistema hematopoyético, hígado, bazo) que suelen presentarse como citopenias (al menos 2 series: hemoglobina <10g/dl, leucocitos <4.000/mm3, trombocitopenia <100.000/mm3), hepatomegalia >3cm y/o esplenomegalia > 2cm del reborde costal o disfunción hepática (hipoproteinemia <5,5g/dl, hipoalbuminemia <2,5g/dl, elevación de la gamma glutamil transpeptidasa o GGT)4–6,9,20–22.
Se recomienda realizar pruebas complementarias analíticas como hemograma, coagulación, ionograma, velocidad de sedimentación, función hepática y renal, proteinograma, metabolismo del hierro, pero los resultados son inespecíficos21. Los estudios radiológicos deben incluir ecografía abdominal, radiología simple, tomografía computarizada (TC) o resonancia magnética (RM) de las lesiones óseas sospechosas y otros huesos20–22. Existe controversia sobre la técnica de elección para valorar lesiones líticas, ya que generalmente se detectan bien en RX simple, pero puede ser difícil en localizaciones como costillas, columna vertebral y pelvis. La TC o la RM son útiles para valorar mejor las lesiones óseas y de partes blandas. El papel del PET-TC con 18-fluorodeoxiglucosa (18F-FDG) no está bien establecido, pero es muy sensible y podría ser útil, en recaídas o casos complejos21,27. Según la sintomatología y la sospecha de órganos afectados se realizarán otros estudios para establecer la afectación de los mismos: estudios hormonales (p. ej., prueba de deprivación hídrica), RM craneoespinal o del eje hipotálamo-hipofisario, audiometría, potenciales evocados sensitivos, evaluación neurocognitiva y psicométrica, TC pulmonar, pruebas funcionales pulmonares, lavado broncoalveolar, gastroscopia, colonoscopia, etc.4–6,20–22.
En el seguimiento de los pacientes se deberán realizar diversas pruebas complementarias, en función de la localización de las lesiones. Es importante conocer que la respuesta radiológica de las lesiones óseas puede tardar meses en evidenciarse y se recomienda mantener el mismo método de evaluación. El seguimiento radiológico suele limitarse a la evaluación de las regiones anatómicas afectadas inicialmente, salvo clínica compatible con nuevas lesiones22,23.
Manejo terapéutico de las formas leves: cutáneas, óseas simples, óseas en localizaciones especiales y otras localizaciones únicasEn general, los pacientes con enfermedad de un solo sistema y confinada a una única localización, solo requieren terapia local u observación después de la biopsia inicial. Los pacientes con lesiones únicas presentan buen pronóstico y pueden resolverse espontáneamente20–23.
Compromiso cutáneo aisladoLas lesiones cutáneas son frecuentes y variadas, aunque la afectación exclusiva en piel es poco común (5%). Aparecen a cualquier edad, pero son más características de recién nacidos y lactantes. Suelen presentarse como eccema seborreico o costra láctea persistente en cuero cabelludo, lesiones en pliegues y periné o exantema generalizado. Pueden presentar pápulas, pústulas, vesículas o úlceras con costras hemorrágicas. En la HCL cutánea exclusiva se recomienda observación y seguimiento estrecho porque pueden desaparecer sin tratamiento. pero también pueden sufrir reactivación o progresión multisistémica hasta en el 40%, especialmente en pacientes con mutación BRAFV600E. En lesiones únicas la resección quirúrgica con fines diagnósticos suele ser suficiente para la curación28–31.
Se recomienda administrar tratamiento solo a pacientes sintomáticos, pero no existen recomendaciones estándar para formas exclusivas cutáneas. Las principales opciones de tratamiento son20–24,32,33:
- •
Corticoesteroides tópicos de potencia media y alta constituyen la terapia de primera línea, pero no siempre son eficaces y es común la recidiva después de interrumpirlos. También se pueden usar tandas cortas por vía oral.
- •
Metotrexato, mercaptopurina o hidroxiurea oral. Se puede administrar metotrexato: 20mg/m2 semanal, mercaptopurina: 50mg/m2/día o hidroxiurea 20mg/kg/día, durante 6 a 12 meses, solos o combinados en casos refractarios. También la talidomida oral (dosis de 50 a 200mg/día) puede mostrar eficacia en niños y adultos.
- •
Otras terapias en formas refractarias: La aplicación tópica de mostaza nitrogenada o imiquimod o psoraleno con radiación ultravioleta B (PUVB) puede ser eficaz y segura en formas resistentes y centros especializados. En enfermedad cutánea extensa y sintomática, se puede administrar quimioterapia sistémica (combinación de esteroides con vinblastina durante 6 a 12 meses).
Las lesiones óseas están presentes en aproximadamente el 80% y pueden presentarse en cualquier hueso del cuerpo, aunque son raras en manos y pies. Los sitios más comúnmente afectados son cráneo, seguido de columna, extremidades y pelvis. Pueden ser asintomáticas o asociar dolor, tumefacción de partes blandas o limitación de la movilidad. Suelen ser lesiones líticas con masa de partes blandas19–23.
Las lesiones óseas únicas (50%) pueden regresar espontáneamente y la observación y seguimiento pueden ser suficientes. Muchas lesiones mejoran después de la biopsia. Para considerar si precisa tratamiento se valorarán los síntomas clínicos, tamaño y localización de la lesión, especialmente la compresión de médula espinal, deformidad inaceptable, dolor intenso y discapacidad funcional. Las opciones de tratamientos dependerán del tipo de lesión19–23,30,34–37:
- -
Lesiones craneales únicas de la calota craneal a nivel frontal, parietal, occipital o en cualquier otro hueso:
- •
Actitud conservadora inicial. Esperar a la regresión espontánea tras la biopsia.
- •
Legrado o curetaje/cirugía. La escisión completa de la lesión ósea pequeña (< 2cm) puede estar indicada al biopsiar. Pero no está indicada la escisión radical de lesiones grandes (> 5cm) porque aumenta el defecto óseo, prolonga el tiempo de curación y podría resultar en defectos esqueléticos permanentes. Para lesiones de 2 a 5cm de diámetro, la biopsia o legrado parcial son suficientes. La inyección intralesional de corticoides en el acto quirúrgico muestra efecto farmacológico prolongado, alivio del dolor y mejora la movilidad con buena tolerancia en lesiones sintomáticas accesibles.
- •
Indometacina oral. Ha demostrado su eficacia como terapia inicial o en recaídas para controlar el dolor, revertir la enfermedad e inducir respuesta en >90% de lesiones óseas sintomáticas, con mínimos efectos secundarios. Dosis recomendada es 1-2mg/kg/día durante una mediana de 6-12 meses35,36.
- -
Lesiones craneales en huesos de la cara, órbitas, apófisis mastoides y hueso temporal. La afectación de huesos faciales y fosas craneales anterior y media (temporal, órbita, esfenoides, etmoides y zigomático) que presentan extensión tumoral intracraneal, comprenden un grupo de riesgo para el sistema nervioso central (SNC) y pueden asociar mayor probabilidad de diabetes insípida y afectación con patrón neurodegenerativo36. Se recomienda quimioterapia sistémica con vinblastina y prednisona (estrato I del protocolo vigente LCH-IV) en general, pero hay cierta controversia ya que algunas lesiones pueden tener evolución favorable con actitud conservadora o indometacina32,35,36.
- -
Lesiones óseas vertebrales. Pueden afectar cualquier vértebra, aunque es más común en cervicales y producir aplastamiento del cuerpo vertebral (vértebra plana). Suelen ser dolorosas y excepcionalmente asocian deficiencias neurológicas por extensión de masa de partes blandas y compresión de médula espinal. La recomendación actual es observación, salvo en caso de masa de partes blandas y extensión intraespinal que pueden requerir quimioterapia. El tratamiento ortopédico con ortesis tipo corsé puede ser necesario cuando hay inestabilidad vertebral o síntomas neurológicos.
El riesgo de reactivación de una lesión ósea única es bajo (10%). En casos de afectación ósea recidivante, resistente o progresiva el tratamiento se basa en indometacina, o quimioterapia (prednisona+vinblastina, citarabina+vincristina, cladribina, hidroxiurea). También algunas recaídas óseas aisladas leves regresan espontáneamente y los bisfosfonatos puede ser eficaces en algunos casos20–23.
Afectación ganglionarLa extirpación quirúrgica es el tratamiento de elección, si hay afectación única32.
Afectación pulmonar aisladaEn adolescentes el abandono del tabaquismo es clave para conseguir la curación. En casos sintomáticos se recomienda quimioterapia.
Lesiones ocupantes de espacio en sistema nervioso centralPueden aparecer en cualquier localización, pero más frecuentemente en región hipotálamo-hipofisaria con engrosamiento del tallo hipofisario. En algunas localizaciones la extirpación quirúrgica puede ser diagnóstica y curativa. Otras lesiones suelen responder a quimioterapia (vinblastina/corticoides, vincristina/ARA-C, cladribina en monoterapia)39.
La diabetes insípida central (DIC), puede preceder a la HCL o aparecer posteriormente. Estos pacientes tienen mayor riesgo de afectación neurodegenerativa, pero las terapias específicas de HCL no consiguen revertir la DIC, por lo que no se recomiendan32,38.
Manejo terapéutico de formas multisistémicas y disfunción orgánicaLos pacientes se clasifican en diferentes categorías de riesgo, según la extensión de la enfermedad y órganos implicados. La afectación de varios sistemas orienta a la necesidad de eliminar los progenitores clonales con quimioterapia sistémica. El tratamiento con corticoides y vinblastina ha demostrado una excelente respuesta en pacientes con enfermedad multisistémica y se propone como tratamiento de primera línea por la Histiocyte Society8,9,31.
Dada la baja prevalencia de la HCL, los estudios multicéntricos han sido fundamentales para conseguir resultados y establecer las recomendaciones terapéuticas. En España destaca la labor del grupo de trabajo de histiocitosis de la Sociedad Española de Hematología y Oncología Pediátricas (SEHOP) y la participación en estudios colaborativos españoles (HCL-95, HCL-2000) e internacionales (LCH-III, LCH-IV). Los resultados publicados de LCH-III en 554 pacientes pediátricos HCL multisistémica (HCL-MS) muestran una supervivencia global del 99% sin órganos de riesgo afectados y del 84% si afectación de órganos de riesgo, mejorando el pronóstico respecto a estudios previos (LCH-I, LCH-II). La adición de metotrexato no aportaba beneficio. Se demostró el beneficio de prolongar la terapia para reducir las recaídas9. Recientemente se ha descrito el impacto pronóstico negativo de la afectación gastrointestinal en HCL-MS37.
El estudio internacional actual LCH-IV (ClinicalTrials.gov: NCT02205762) incluye recomendaciones para todas las formas de HCL, incluyendo casos refractarios y recidivantes. El promotor en España es SEHOP, participan 27 centros, coordinados desde Instituto Investigación Sanitaria BioCruces Bizkaia, con apoyo de las plataformas ECLIM-SEHOP y SCReN. Desde 2016, se han incluido 103 pacientes de 23 hospitales.
A pesar de los buenos resultados de supervivencia global, más del 40% de los pacientes con HCL-MS serán refractarios al tratamiento inicial o presentarán recidivas, precisando otras terapias. Se han utilizado diferentes tratamientos de rescate como citarabina, cladribina, clofarabina y trasplante de precursores hematopoyéticos. Desde la identificación de mutaciones en la vía MAPK/ERK se han ampliado las estrategias terapéuticas: como terapia dirigida y fármacos inhibidores con altas tasas de respuesta en niños con HCL de alto riesgo refractaria4–7,39.
Diversas publicaciones recogen la experiencia del tratamiento con vemurafenib y otros inhibidores (dabrafenib, cobimetinib, trametinib), destacando las excelentes tasas de respuesta y su rapidez en formas graves7,10,11,32,39. Sin embargo, esta terapia no es curativa porque las recaídas son casi constantes tras su suspensión. Los ensayos clínicos deberían confirmar su eficacia y seguridad, la duración y momento óptimo para su uso y la utilidad de las terapias combinadas de inhibidores de la vía MAPK/ERK con quimioterapia.
Seguimiento: secuelas a largo plazo y neurodegeneraciónLas reactivaciones, afectación de órganos de alto riesgo, secuelas a largo plazo y la enfermedad neurodegenerativa asociada a la HCL representan los mayores desafíos diagnósticos y terapéuticos. Alrededor del 50% de los pacientes con HCL presenta algún tipo de secuela, sobre todo en HCL-MS. Las reactivaciones y lesiones de riesgo SNC asocian mayor tasa de complicaciones a largo plazo. Las secuelas aparecen más frecuentemente en hueso, piel y área hipotálamo-hipofisaria, aunque pueden afectar a otras localizaciones22,38.
Las secuelas ortopédicas (secundarias a fracturas patológicas de huesos largos, deformidades, escoliosis) son frecuentes en lesiones óseas (20-30% de pacientes, sobre todo en menores de 3 años). La afectación de huesos craneales puede provocar proptosis, asimetrías faciales, pérdida de audición, pérdida de piezas dentarias, etc.22,35,38. Las lesiones cutáneas pueden provocar ulceraciones, cicatrices y secuelas cosméticas28,29.
La afectación del eje hipotalámico-hipofisario puede producir secuelas endocrinológicas permanentes como diabetes insípida, que no responden al tratamiento de HCL. Las deficiencias hormonales de la hipófisis anterior son menos habituales, pero provocarán retraso del crecimiento, hipotiroidismo, hipogonadismo, amenorrea, pubertad precoz o retrasada38.
La afectación neurodegenerativa (HCL-ND) se caracteriza por un síndrome progresivo de alteraciones radiológicas y manifestaciones clínicas, con escasa correlación clínico-radiológica. Es un síndrome tardío y progresivo caracterizado por la afectación cerebelosa y del tronco encéfalo que puede asociar deterioro neurocognitivo, que ocurre habitualmente entre 5-15 años después del diagnóstico inicial. La incidencia varía desde el 1 al 24%, dependiendo de factores de riesgo identificados como la afectación de huesos del macizo facial, presencia de DIC y mutación de BRAFV600E que aumentan la probabilidad. El diagnóstico es complejo y las recomendaciones terapéuticas todavía no están bien establecidas. Se han planteado diversas terapias con inmunomoduladores y citostáticos con mala respuesta y se podría considerar el uso de inhibidores de MAPK en determinados casos40.
FinanciaciónEste trabajo ha sido financiado parcialmente por los fondos del Proyecto de Investigación sobre Histiocitosis de la Fundación Vasca de Innovación e Investigación Sanitaria BIOEF (BIO16/ER/020/BC) y de la Asociación Española contra la Histiocitosis-ACHE (BC/A/15/012, BIOEF11/017, BIOEF09/047) cuya investigadora principal es Itziar Astigarraga.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los pacientes, familiares y profesionales que han participado en los estudios colaborativos de histiocitosis. A Ana Barriuso por su ayuda técnica en la elaboración del documento. A Marta Llarena, Ane Karmele Sáenz de Arzamendi y a la plataforma SCReN (Spanish Clinical Research Network) del Instituto de Salud Carlos III por su labor de apoyo y monitorización del estudio internacional LCH-IV. A la plataforma ECLIM-SEHOP por su ayuda en la implementación y el desarrollo de los ensayos clínicos internacionales multicéntricos en cáncer infantil en España.
