


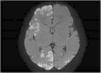
A female full-term neonate developed clonic seizures in the left side of the body in the first day of life accompanied by eye deviation. The seizures were refractory to first-line anticonvulsant therapy and resolved with phenytoin. The brain MRI revealed diffuse hyperintense cortical lesions with an extensive, bilateral and patchy distribution at the periventricular and subcortical levels (Fig. 1). The EEG, infectious and metabolic disease tests and ophthalmological evaluation were normal.

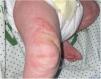
On day 9, she developed a rash consisting of papulovesicular plaques in the upper and lower extremities and back, without a dermatomal distribution (Fig. 2). The mother was interviewed again, revealing a maternal history of hypopigmented cutaneous lesions, dental and ungual anomalies, strabismus, supernumerary nipple, previous miscarriages and convulsive seizures of unknown etiology in the neonatal period.

Due to suspected genetic neurocutaneous disease, genetic testing was ordered that identified a mutation in the IKBKG gene, confirming the diagnosis.
Incontinentia pigmenti is an X-linked genodermatosis. It affects tissues derived from the ectoderm. Neurologic involvement is frequent (30–50%), but it is rarely the initial manifestation.1
It is an underdiagnosed disease and it may be confused with other conditions, such as congenital infections, hypoxic-ischemic encephalopathy, metabolic disorders, hereditary epidermolysis bullosa… Early diagnosis is important, as neonatal neurologic and ophthalmologic involvement are indicators of a poor prognosis and may affect neurodevelopment.2,3
