



Anomalous origin of the pulmonary artery from the ascending aorta (AOPA), also known as hemitruncus, is a rare condition that accounts for 0.3% of cases of congenital heart disease.1 Right-sided AOPA (AORPA) is more frequent than left-sided AOPA (AOLPA). The anomaly produces progressive pulmonary vascular obstructive disease and pulmonary hypertension (PH).2 Thus, early diagnosis is paramount to allow prompt surgical repair and to reduce morbidity and mortality.2 We present four cases of AOPA in pediatric patients.
Patient 1 had a prenatal diagnosis of tetralogy of Fallot and right aortic arch. The angiography at 2 weeks post birth ordered for evaluation of persistent heart failure (HF) confirmed the presence of AOLPA, which was missed in the initial echocardiogram. He also had an anorectal malformation and vesicoureteral reflux. Genetic testing identified a 22q11 microdeletion. The patient underwent reimplantation of the affected pulmonary artery via direct anastomosis to the main pulmonary artery with concomitant repair of tetralogy of Fallot. At present, 18.1 years after surgery, the patient is asymptomatic and has mild right ventricular outflow tract obstruction.
Patient 2 had a prenatal diagnosis of interrupted aortic arch type B and ventricular septal defect. She had HF at 2 days post birth. The postnatal CT scan revealed AORPA, which was missed in the initial echocardiogram (Fig. 1A–G). She was found to carry a 22q11 microdeletion. Surgery was not performed in accordance with the parents’ wishes, due to the high complexity of the congenital heart disease in the context of her genetic mutation. Life sustaining treatment was withheld. She died three days after birth.

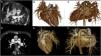
Computed tomography images of patient 2.
(A–G) Left pulmonary artery (LPA) originating from the main pulmonary artery (PA). (D–G) Patent ductus arteriosus (PDA) communicating the PA with the descending aorta (DA). (E–G) Volume-rendered images show the anomalous origin of the RPA from the ascending aorta (AAo) with an interrupted aortic arch. (C, E, G) Small right pulmonary artery (RPA) (red arrow) originating from the ascending aorta (AAo).
Patient 3 received a diagnosis of isolated AORPA at age 2 months following the detection of a heart murmur (Fig. 2A–C). Genetic testing was negative. He underwent reimplantation of the affected pulmonary artery via direct anastomosis to the main pulmonary artery. He developed progressively severe pulmonary stenosis of the reimplanted right pulmonary artery that required angioplasty with stent implantation 7.4 years after surgery. He is currently asymptomatic with no residual lesions, 7.9 years after surgical intervention.

Patient 4 presented at 2 months of age with HF. The echocardiogram revealed isolated AORPA, which was confirmed by CT (Fig. 2A–C). Genetic testing was negative. He underwent reimplantation of the affected pulmonary artery with direct anastomosis to the main pulmonary artery. He is currently asymptomatic with no residual lesions, 5.4 years after surgery.
In agreement with previous reports, our patients presented with HF secondary to the left-to-right shunt due to the high pulmonary flow to both lungs.2 Thus, these patients are at increased risk of developing early PH and progressive pulmonary vascular obstructive disease.2 Consistently, all our patients presented with PH.
A few cases of AOLPA associated with chromosome 22q11 microdeletion have been described in the literature.3 Two of our patients had this microdeletion. Saliba et al. argued that this association demonstrates that AOLPA belongs to the spectrum of conotruncal heart malformations resulting from neural crest maldevelopment.3
Recently, eight cases of prenatal diagnosis of AOPA have been reported.4 However, making a precise diagnosis of AOPA through fetal echocardiography remains challenging. In this regard, none of our patients were prenatally diagnosed. Additionally, although echocardiography is the first-line imaging technique, it might not achieve diagnosis in 15% of cases.5 Therefore, cardiac catheterization and CT are required for diagnosis confirmation and pulmonary vascular resistance assessment.1 Concordantly, echocardiography failed to diagnose 50% of our patients, requiring the aforementioned imaging techniques for its diagnosis. Our higher rate of missed diagnosis could be related to our small sample size, the infrequency of this condition and/or its low awareness among clinicians.
Prompt surgical repair of AOPA, preferably within the neonatal period, is preferred to prevent pulmonary vascular obstructive disease, which can rapidly evolve after 3 months of age.6
Prompt surgical intervention achieves good survival and hemodynamic outcomes.2 All our patients underwent intervention before age 3 months and none of them had developed PH by the end of the study period. While there are multiple surgical techniques, direct anastomosis of the anomalous pulmonary branch to the main pulmonary artery trunk is the preferred and most effective and widely used technique.1,2 This approach was also the established approached in our hospital (Fig. 2D–F). Alternative surgical techniques involve reimplantation with pericardial patch augmentation or homograft interposition when the distance between the anomalous pulmonary artery and the main pulmonary artery is too long for direct anastomosis.
In conclusion, it is essential that pediatricians consider AOPA in the differential diagnosis of infants with HF, as timely recognition and surgical correction are crucial to prevent irreversible pulmonary vascular disease and the development of PH, as demonstrated in our study. In this regard, it is crucial to verify the origin of the pulmonary arteries through fetal echocardiography. Additional studies with long-term follow-up are needed to confirm our findings and further define this entity to increase clinical awareness and facilitate early diagnosis.
CRediT authorship contribution statementThe authors assert that all procedures contributing to this work complied with the ethical standards of the relevant national guides and have been approved by the institutional committee of Hospital Sant Joan de Déu.
FundingThis research did not receive any external funding.
The authors have no conflicts of interest to declare.
