



El origen anómalo de una de las ramas de la arteria pulmonar (OAAP), también conocido como hemitruncus, es una malformación rara que representa el 0,3% de los casos de cardiopatía congénita1. El origen anómalo de la arteria pulmonar derecha (OAAPD) es más frecuente que el de la izquierda (OAAPI). Estas anomalías producen enfermedad pulmonar obstructiva crónica e hipertensión pulmonar (HP)2. Por lo tanto, su diagnóstico temprano es crucial para permitir la intervención quirúrgica precoz y reducir la morbimortalidad2. Se presentan cuatro casos de OAAP en pacientes pediátricos.
El primer caso corresponde a un paciente con diagnóstico prenatal de tetralogía de Fallot y arco aórtico derecho. La angiografía a las 2semanas de vida, indicada por insuficiencia cardíaca (IC) persistente, identificó OAAPI, que había pasado desapercibida en la ecocardiografía inicial. También presentaba una malformación anorrectal y reflujo vesicoureteral. En el estudio genético se detectó microdeleción 22q11. Se practicó reimplante de la arteria pulmonar afectada al tronco de la arteria pulmonar mediante anastomosis directa, con corrección concomitante de la tetralogía de Fallot. Actualmente, a los 18,1años de la cirugía, se encuentra asintomático, con obstrucción leve del tracto de salida del ventrículo derecho.
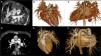
El segundo caso correspondió a una paciente con diagnóstico prenatal de interrupción del arco aórtico tipoB y comunicación interventricular. A los dos días de nacer presentó IC. La TC postnatal reveló OAAPI, que había pasado desapercibida en la ecocardiografía inicial (fig. 1A-G). Se detectó microdeleción 22q11. De acuerdo con el deseo de los padres, no se realizó intervención quirúrgica por la alta complejidad de la cardiopatía congénita en el contexto de la mutación genética presente, decidiéndose limitar el esfuerzo terapéutico. La paciente falleció a los tres días de vida.

Imágenes de tomografía computarizada del caso2. A-G)Arteria pulmonar izquierda (API) con origen en el tronco de la arteria pulmonar (AP). D-G)Ductus arterioso persistente (DAP) que comunica la arteria pulmonar con la aorta descendente (AoD). E-G)Reconstrucciones tridimensionales que muestran el origen anómalo de la APD desde la aorta ascendente (AoA) y la interrupción del arco aórtico. C,E,G)Arteria pulmonar derecha (APD) pequeña (flecha roja) con origen en la aorta ascendente (AoA).
El tercero correspondió a un paciente diagnosticado de OAAPD aislado a los dos meses de edad tras la detección de un soplo cardíaco (fig. 2A-C). El estudio genético fue negativo. Se practicó reimplante de la arteria pulmonar afectada al tronco de la arteria pulmonar mediante anastomosis directa. El paciente desarrolló estenosis pulmonar progresivamente grave en la arteria pulmonar derecha reimplantada que requirió angioplastia con implantación de stent a los 7,4años de la intervención. Actualmente, a los 7,9años de la cirugía, se encuentra asintomático y sin lesiones residuales.

Imágenes de tomografía computarizada del caso4. A)Arteria pulmonar izquierda (API) con origen en el tronco de la arteria pulmonar (AP). Arteria pulmonar derecha (APD) con origen en la aorta ascendente (AoA). B-C)Reconstrucciones tridimensionales. D-F)TC de control tras la intervención quirúrgica.
En el cuarto caso, el paciente presentó IC a los dos meses de edad. La ecocardiografía reveló OAAPD aislado, confirmado por TC (fig. 2A-C). El estudio genético fue negativo. Se practicó reimplante de la arteria pulmonar afectada al tronco de la arteria pulmonar mediante anastomosis directa. Actualmente, a los 5,4años de la cirugía, se encuentra asintomático y sin lesiones residuales.
En consonancia con la literatura previa, nuestros pacientes presentaron IC secundaria al cortocircuito izquierda-derecha por el elevado flujo pulmonar en ambos pulmones2. Por lo tanto, estos pacientes presentan un mayor riesgo de HP precoz y progresión a enfermedad vascular pulmonar obstructiva2. De este modo, todos nuestros pacientes presentaron HP.
Se han descrito pocos casos de OAAPI asociados a microdeleción 22q113. Esta microdeleción se identificó en dos de nuestros pacientes. Saliba et al. sostienen que esta asociación demuestra que el OAAPI pertenece al conjunto de malformaciones cardíacas conotruncales debidas a un desarrollo anómalo de la cresta neural3.
Recientemente se han descrito ocho casos de diagnóstico prenatal de OAAP4. A pesar de ello, el diagnóstico preciso de la OAAPI mediante ecocardiografía fetal sigue siendo complejo. En la presente serie, por ejemplo, no se diagnosticó prenatalmente en ninguno de los pacientes. Además, aunque la ecocardiografía es la prueba de imagen inicial, la anomalía puede pasar desapercibida en hasta el 15% de los casos5. Por ello, se requiere la realización de cateterismo cardíaco y tomografía computarizada para confirmar el diagnóstico y valorar las resistencias vasculares pulmonares1. Así, la ecocardiografía no detectó la anomalía en el 50% de nuestros pacientes, cuyo diagnóstico requirió el uso de las técnicas de imagen ya mencionadas. La mayor tasa de diagnósticos erróneos en nuestra serie podría estar relacionada con el reducido tamaño de la muestra, la escasa incidencia de esta afección y/o la baja concienciación de los profesionales.
La corrección quirúrgica precoz del OAAP, preferiblemente en el periodo neonatal, es la mejor opción para prevenir la enfermedad obstructiva vascular pulmonar, que puede desarrollarse rápidamente a partir de los tres meses de edad6.
La intervención quirúrgica precoz se asocia a una tasa de supervivencia elevada y resultados hemodinámicos favorables2. En nuestra serie, todos los pacientes fueron intervenidos antes de los tres meses de edad y ninguno había desarrollado HP al final del estudio. Aunque existen múltiples técnicas quirúrgicas, la anastomosis directa de la rama pulmonar anómala al tronco de la arteria pulmonar es la técnica preferida, la más eficaz y la más utilizada1,2. También es la preferida en nuestro centro (fig. 2D-F). Entre las alternativas quirúrgicas se encuentran el reimplante con ampliación con parche pericárdico o interposición de un homoinjerto cuando la distancia entre la arteria pulmonar anómala y el tronco de la arteria pulmonar es excesiva para realizar una anastomosis directa.
En conclusión, es fundamental que los pediatras consideren la OAAP en el diagnóstico diferencial de la insuficiencia cardíaca en lactantes, ya que el reconocimiento precoz y la corrección quirúrgica temprana son cruciales para prevenir la enfermedad vascular pulmonar irreversible y el desarrollo de HP, tal como se ha observado en nuestra serie. En este sentido, resulta esencial confirmar el origen de las arterias pulmonares mediante ecocardiografía fetal. Se precisan estudios a largo plazo que permitan confirmar estos resultados y definir mejor esta entidad con objeto de aumentar la concienciación entre los profesionales y favorecer su diagnóstico precoz.
FinanciaciónEste trabajo no ha recibido financiación externa.
Consideraciones éticasLos autores declaran que todos los procedimientos realizados en el marco de este trabajo cumplen las normas éticas de las guías nacionales vigentes y cuentan con la aprobación del comité de ética de investigación del Hospital Sant Joan de Déu.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
