


La microduplicación 3q29 (MIM 611936) es un raro síndrome caracterizado por retraso mental moderado, rasgos dismórficos craneofaciales y anomalías musculoesqueléticas. La región mínima crítica tiene un tamaño de aproximadamente 1,73Mb, está flanqueada por secuencias repetitivas y es similar en tamaño a la microdeleción recíproca 3q29, sugiriendo para ambas una recombinación homóloga no alélica (NAHR) entre las secuencias repetitivas como mecanismo de producción. Describimos una nueva familia con diferente expresividad clínica en la paciente y su madre.
3q29 microduplication (MIM 611936) is rare syndrome characterized by moderate mental retardation, craniofacial dysmorphic features and musculoskeletal anomalies. The size of the minimal critical region is about 1.73Mb. It is flanked by repetitive sequences and it is similar in size to the reciprocal 3q29 microdeletion, suggesting a non-allelic homologous recombination event (NAHR) at flanking LCR sequences as its aetiological mechanism. We describe a new familial case with variable expressivity.
La reciente introducción de una potente tecnología que permite realizar un cribado de todo el genoma (array-CGH) para detectar cambios en el número de copias (copy number variation [CNV]) (microdeleciones/microduplicaciones) responsables de una gran variedad de manifestaciones clínicas, incluidos el retraso mental y las malformaciones congénitas, y con un poder de resolución muy superior al de las técnicas citogenéticas convencionales, ha supuesto no solo un notable incremento diagnóstico desde un punto de vista cuantitativo (se detectan CNV patogénicos en el 15-20% de los casos con estudio convencional normal)1, sino también un aumento en la descripción de nuevos síndromes, como es el caso de la microdeleción (MIM 609425)/microduplicación (MIM 611936) 3q292–4.
Estos síndromes de microdeleción/microduplicación tienen un mecanismo común de producción: en el genoma humano existen diversos tipos de secuencias repetitivas, entre las que encontramos las llamadas duplicaciones segmentarias (segmentary duplications [SD]) de tamaño > 1Kb y caracterizadas por un grado alto de similitud de secuencia (96-100%). Estas duplicaciones segmentarias pueden dar lugar a fenómenos de recombinación meiótica aberrante (non-allelic homologous recombination [NAHR]) que conducen a la deleción/duplicación de los segmentos genómicos comprendidos entre dos SD5,6. Este mecanismo de producción se ha descrito para ciertos síndromes, como por ejemplo neuropatía hereditaria sensible a la presión (deleción de 1,5Mb en 17p11.2)/Charcot-Marie-Tooth tipo 1A (duplicación de 1,5Mb en 17p11.2).
Aunque un mismo mecanismo (NAHR) puede explicar la formación de los síndromes de microdeleción/microduplicación, los segundos son diagnosticados con menos frecuencia. Esto se debe a un sesgo diagnóstico por presentar un fenotipo más leve y variable y por las limitaciones de las técnicas citogenéticas convencionales.
Caso clínicoSegunda hija de padres jóvenes, sanos y no consanguíneos. Un hermano de 3 años, sano. Embarazo natural sin incidencias. Parto espontáneo en la semana 37, vaginal, cefálico. Apgar de 8/9. Peso 2.650g, longitud 51cm, perímetro craneal 33cm. Se detectó soplo cardiaco sistólico con ECG normal y pulsos femorales palpables. A los 2meses se diagnosticó de CIV perimembranosa de 4mm con cortocircuito izquierda-derecha, estenosis valvular pulmonar con gradiente de 30mmHg, foramen ovale con mínimo cortocircuito y presión pulmonar normal. A los 3años consulta por retraso del lenguaje, apreciándose rasgos dismórficos (fig. 1): sinofridia (posteriormente corregida estéticamente, no visible en la fotografía) y pestañas largas, cejas pobladas, narinas antevertidas, orejas de implantación baja, labio superior fino, filtrum largo, micromelia, pulgar de implantación proximal, paladar ojival, hipoplasia de labios menores. Se plantearon los siguientes diagnósticos: Turner mosaico, Floating-Harbor, microdeleción 22q11.2, Cornelia de Lange. A los 5años: RM de hipófisis, hipotálamo y sistema nervioso central, sin hallazgos anormales. Edad ósea de 3años. Minusvalía legal del 33%. A los 11 años y medio presenta retraso del desarrollo con retraso escolar, necesitando apoyo. No ha sido intervenida de su CIV. La madre presenta un trastorno de la motilidad ocular con nistagmus rotatorio bilateral de pequeño grado. No se observan otras manifestaciones clínicas.
Estudios genéticos
El estudio citogenético convencional (nivel de bandeo GTG550) fue normal: cariotipo 46,XX. Posteriormente, se realizó un microarray-CGH Cytoarray (44K) (Agilent Technologies, Santa Clara, CA, EE: UU.), que detectó una duplicación en la citobanda 3q29 de 1,73Mb (chr3: 197266316-198996325) (fig. 2). Esta microduplicación, no detectable con técnicas citogenéticas convencionales, contiene múltiples genes entre los que cabe destacar: TFRC, ZDHHC19, PAK2 y DLG1. En el microarray se detectan otros cambios de número de copia descritos en las bases de datos como polimorfismos (duplicación 1p36.33 de 0,002Mb y deleción 1q21.1 de 0,049Mb). El estudio familiar demuestra que la duplicación es de origen materno.
Discusión
La microduplicación 3q29 (MIM 611936) fue descrita por primera vez en una familia con retraso mental leve y rasgos dismórficos2. Nuestro caso y los descritos en la bibliografía definen una región mínima crítica, aproximadamente de 1,6Mb comprendida entre los genes TFRC y BDH1 y flanqueada por duplicaciones segmentarias agrupadas. Esta región contiene 20genes, de los cuales hay tres de función desconocida y cuatro relacionados con patología: DLG1, PAK2, TFRC y ZDHHC19. El gen DLG1 es un buen candidato para las anomalías oculares, ya que se expresa en el cristalino en desarrollo y en el epitelio pigmentario retiniano. Pertenece a la superfamilia de genes MAGUK que intervienen en un gran número de funciones celulares como la regulación de la proliferación celular, polaridad celular, sinaptogénesis, la organización del citosqueleto y la elongación morfológica del embrión7. El retraso en el desarrollo podría estar relacionado con los genes PAK2 y DLG1 al ser homólogos de los genes PAK3 y DLG3, que se encuentran relacionados con el retraso mental ligado al cromosoma X. PAK2 pertenece a la superfamilia de genes proteincinasa e interviene en el desarrollo dendrítico de las neuronas corticales embrionarias8. TFRC codifica el receptor de la transferrina que interviene en la homeostasis del hierro pero también en el control de la proliferación y el tamaño celular9. El gen ZDHHC19 es homólogo del ZDHHC9, que codifica para la palmitoiltransferasa, cuyas mutaciones causan retraso mental ligado al cromosoma X con hábito marfanoide (OMIM 300646)10. ZDHHC19 es miembro de una familia de genes que codifican palmitoilaciltransferasas. Estas enzimas producen una modificación postranslacional (palmitoilación) que es importante para la actividad, estabilidad, tráfico y localización de ciertas lipoproteínas de membrana11,12. Otro gen localizado en el intervalo es FBXO45, con expresión en el sistema nervioso y fundamental para el desarrollo neuronal y la actividad sináptica13,14.
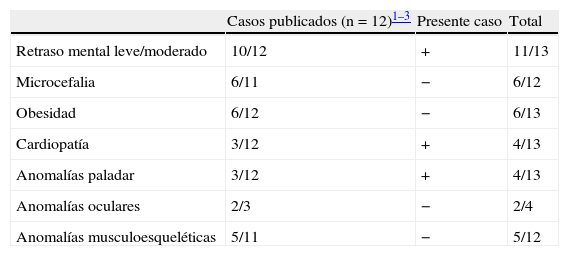
Hasta el momento, sólo 3 autores han comunicado pacientes afectados de microduplicaciones 3q292–4. Ballif describió 19casos y en 5 de ellos la microduplicación era recíproca de la misma región que se encuentra en los síndromes de microdeleción 3q29. De 10casos en los que se estudió a los padres, sólo en dos la microduplicación era de novo. El fenotipo de la microduplicación 3q29 es muy variable, aunque en el conjunto de las tres series publicadas los signos clínicos más frecuentes fueron: retraso mental de leve/moderado, hipotonía, retraso del lenguaje, microcefalia, paladar ojival y defectos cardíacos ventriculares (tabla 1).
Sumario de las manifestaciones clínicas más frecuentes en la microduplicación 3q29
| Casos publicados (n=12)1–3 | Presente caso | Total | |
| Retraso mental leve/moderado | 10/12 | + | 11/13 |
| Microcefalia | 6/11 | − | 6/12 |
| Obesidad | 6/12 | − | 6/13 |
| Cardiopatía | 3/12 | + | 4/13 |
| Anomalías paladar | 3/12 | + | 4/13 |
| Anomalías oculares | 2/3 | − | 2/4 |
| Anomalías musculoesqueléticas | 5/11 | − | 5/12 |
Como observamos en otros casos de microdeleciones/microduplicaciones recíprocas, el cuadro clínico de la microdeleción 3q29 está mejor definido al haberse diagnosticado y descrito más casos que la microduplicación recíproca3. Esto es debido a que, en general, las manifestaciones clínicas producidas por la microdeleción son más graves que las producidas por la microduplicación, al tolerarse peor la haploinsuficiencia de un gen que su duplicación. Mientras que se acepta que la haploinsuficiencia, y por consiguiente, la expresión disminuida de una determinada proteína, tiene un efecto deletéreo sobre una estructura o una ruta metabólica, los mecanismos por los cuales la triple dosis de un determinado gen produce tal efecto son peor conocidos. Cabe esperar que el exceso de dosis de ciertas proteínas que, como es el caso descrito, tienen amplia expresión en el sistema nervioso en formación conduzca a una neurogénesis alterada y, por tanto, produzca manifestaciones clínicas.
En nuestro hospital, la reciente disponibilidad de esta técnica nos ha permitido realizar el diagnóstico molecular en un 16% de casos neuropediátricos analizados con estudio citogenético convencional normal. Debido al mayor rendimiento diagnóstico de la técnica array-CGH frente a las técnicas convencionales se ha descrito recientemente un nuevo protocolo1, en el que se considera el array como prueba diagnóstica inicial. Los estudios de coste-efectividad realizados para esta técnica así lo recomiendan al combinar su mayor capacidad diagnóstica con la eliminación de técnicas complementarias (cariotipo de alta resolución, regiones subteloméricas, etc.)15.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
