




Valproate-induced hyperammonaemic encephalopathy (VHE) is an unusual and serious complication of valproate (VA) treatment. When an early diagnosis is made, it can be reversed with VA withdrawal and early treatment for hyperammonaemia. We describe the case of a 20-day-old male, who developed a serious VHE after receiving VA for refractory neonatal seizures. The VHE was resolved with VA withdrawal in association with carglumic acid and other measures for hyperammonaemia treatment.
La encefalopatía hiperamoniémica inducida por ácido valproico (EHV) es una entidad grave e inusual. Para su diagnóstico, precisa un elevado índice de sospecha, pues resulta reversible con la retirada del fármaco y el tratamiento precoz de la hiperamoniemia. Presentamos el caso de un neonato tratado con valproico (AV) por convulsiones refractarias, que desarrolló una EHV grave que revirtió con la retirada del AV y el tratamiento con ácido carglúmico, junto con otras medidas para control de la hiperamoniemia.
Valproic acid (VA) is an anticonvulsant that, while not being the first-line treatment for convulsions in neonates, is used as a second- or third-line treatment in Spain for convulsions refractory to phenobarbital treatment.1 Valproate-induced hyperammonaemic encephalopathy (VHE) is rare (<1/10,000), but it is serious and potentially fatal.2,3 The main physiopathological mechanism that leads to VHE is a gradual elevation of serum ammonia levels, leading to a clinical syndrome characterised by vomiting, progressive impairment of consciousness up to coma, focal neurological deficits, and increased seizure frequency.4 There is no evidence that the incidence and severity of VHE are associated to blood VA levels, since in most published cases the levels were within therapeutic ranges, although there is evidence suggesting that anticonvulsant polytherapy (phenobarbital, phenytoin, and VA) may contribute to VHE.5
Treatment consists of discontinuation of VA and management of ammonia levels to prevent neurotoxicity, especially in neonates.6 N-carbamylglutamate (NCG), also known as carglumic acid, is a synthetic analogue of N-acetylglutamate (NAG), one of the cofactors essential to the proper functioning of the urea cycle. NCG is indicated for treatment of hyperanaemia secondary to N-acetylglutamate-synthetase (NAGS) deficiency, although it has also been used to treat hyperanaemia of different aetiologies, such as VHE.7
We present the case of a neonate with VHE treated successfully with NCG and other measures against hyperammonaemia.
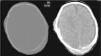
Clinical caseA male neonate, 20 days old, was admitted for a skull fracture and subdural haematoma secondary to occipital trauma, with no family history of metabolic disorders or consanguinity. He had been born at full term in a normal delivery and had an Apgar score of 9/10. The general examination at admission found a left parietal cephalohaematoma and neurological examination found only increased somnolence (Glasgow Coma Scale score 13/15). A computer tomography (CT) scan of the head revealed multiple diastatic skull fractures, extensive right subdural haematoma, and subgaleal fluid collections in the left parietal region (Fig. 1). At 24h after admission, he had partial seizures that were resolved with phenobarbital (initial bolus 20mg/kg, maintenance 5mg/kg/day) and midazolam. He had a new episode that consisted of a prolonged secondarily generalised partial seizure, so VA was added to the treatment regimen (initial bolus 15mg/kg, maintenance 1mg/kg/h). The electroencephalograph (EEG) at admission was normal, but after the seizures it detected focal activity in posterior regions of the right hemisphere that coincided with the location of the subdural haematoma (Fig. 2). At 48h after initiation of VA therapy, the patient started showing signs of decreased level of consciousness, decreased spontaneous movement, and marked hypotonia and hyporeflexia. Examination did not reveal signs of intracranial pressure and a surveillance CT scan of the head did not show any changes. A new EEG showed generalised low-voltage slow activity compatible with cerebral oedema (Fig. 2B). Phenobarbital levels were normal and VA levels were slightly elevated (104.9μg/mL). Basic laboratory testing (complete blood count, electrolyte panel, liver function) yielded normal results, and gasometry showed respiratory alkalosis. The serum ammonia level was 398μmol/L. In light of these findings, treatment with VA was discontinued and treatment with carglumic acid (loading dose 100mg/kg, maintenance 25mg/kg/6h orally) and carnitine (25mg/kg/6h orally) was initiated. Five hours later the level of ammonia had not decreased fast enough (374μmol/L), so oral phenylbutyrate (125mg/kg/6h) and oral L-arginine (600mg/kg/day) were added to the regimen. At 10h, the ammonia level had dropped by half (182μmol/L) and the VA level was within the normal range (69.8μg/mL). At 18h, the level of ammonia was normal (102μmol/L) and the patient had improved considerably, so medication for hyperammonaemia was discontinued. A broad metabolic screening was done on a heel prick blood sample, the results of which were normal. The patient was monitored on an outpatient basis and evolved favourably. Phenobarbital treatment was discontinued at 4 months of age, and at age 1 year he showed normal psychomotor development, with no further episodes of convulsions.
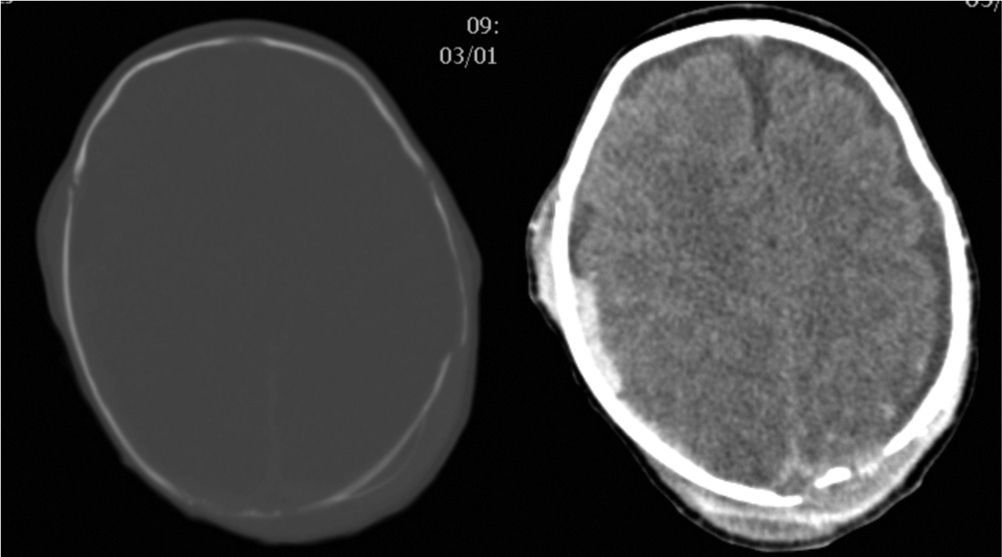
EEG showing focal epileptiform activity in posterior regions of the right hemisphere on an integrated background pattern (A). EEG showing very low voltage diffuse activity formed by a combination of slow frequencies with no clear topographic differentiation or response to external stimuli. The tracing shows focal epileptiform activity in the right posterior temporal region (B).
Although the treatment of neonatal convulsions has hardly changed in the past few decades, it remains a controversial subject because there are no evidence-based guidelines for their management. There is a general consensus that phenobarbital is the first-line treatment,8 followed by phenytoin and benzodiazepines. Instances of treatment with other drugs, such as topiramate or levetiracetam, are starting to be published.9 Valproate is also used as a second-line treatment after phenobarbital treatment failure with positive results, although it should be used with caution because there is limited experience in its use in neonatology.1,10
VHE is a rare and serious condition, especially in neonates. The main physiopathological mechanism involved is an increased serum ammonia level that causes an acute or subacute clinical presentation characterized by vomiting and decreasing level of consciousness that progresses towards coma and that may be associated to focal neurological deficits and increased seizure frequency.4
Ammonia is highly toxic to the central nervous system, as it diffuses freely through the blood–brain barrier, reaching concentrations higher than those found in the blood. The developing brain is much more susceptible to the harmful effects of ammonia than a mature brain.11 Its toxicity is associated to the elevation of extracellular glutamate levels and the development of cerebral oedema with increased intracranial pressure.4 The urea cycle is the main metabolic pathway to eliminate ammonia, transforming it into urea that is later excreted by the kidney.
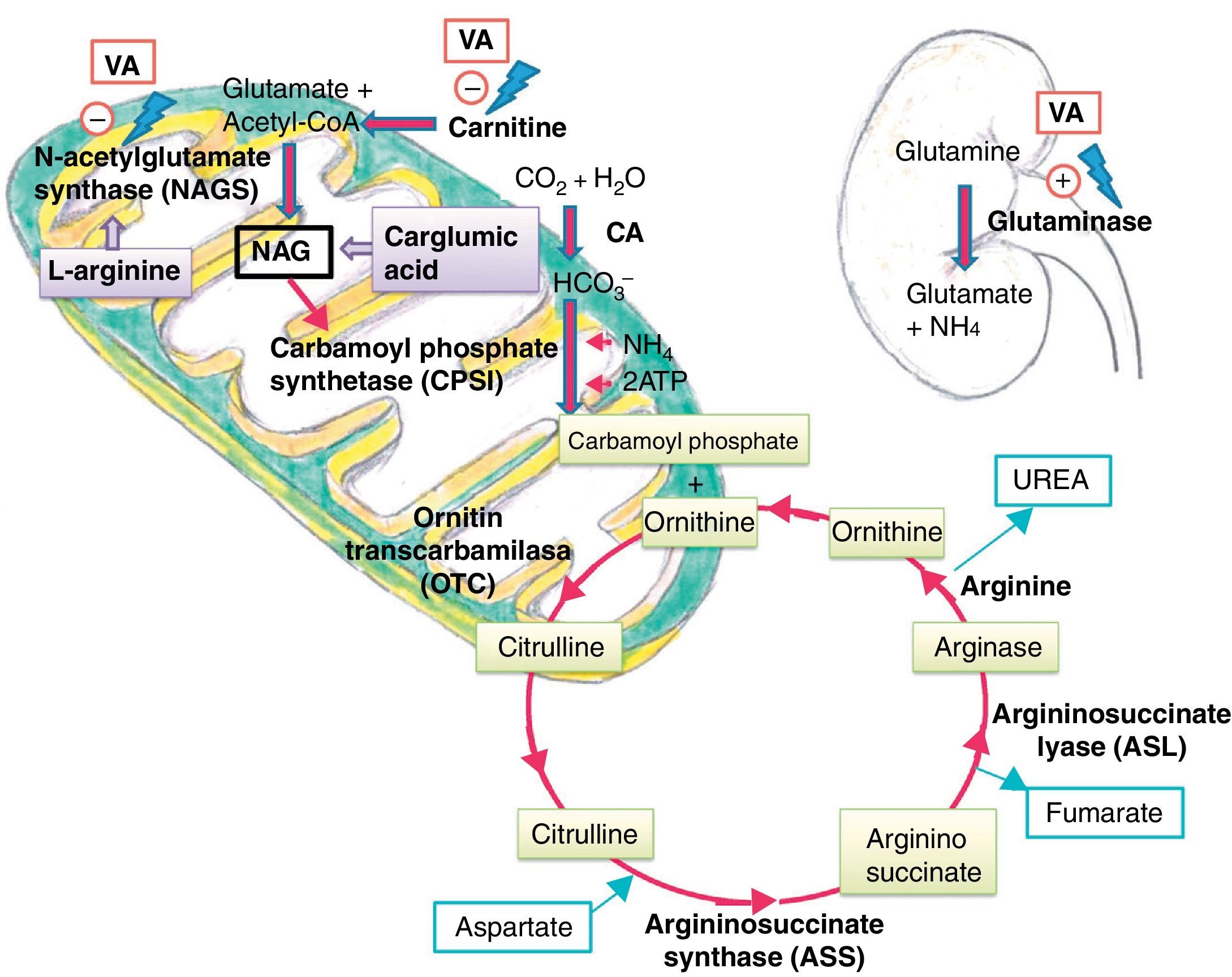
Several mechanisms have been postulated to explain how VA and its metabolites cause hyperammonaemia. One is direct action in the kidney by stimulation of renal glutaminase, and indirect action in the liver by inhibition of NAGS, which reduces the availability of NAG, a cofactor required for the activity of carbamoyl phosphate synthetase (CPS I), the first enzyme in the urea cycle.5 They also cause carnitine depletion, affecting the beta-oxidation of fatty acids and resulting in decreased levels of acetyl-CoA, which is necessary for NAG synthesis11 (Fig. 3).
Although a positive correlation between ammonia levels and the dosage and serum levels of AV has been reported recently,12 a significant association between the daily dosage of VA and the development and severity of VHE has not been found, in fact the levels of VA are within therapeutic5 or supratherapeutic2 ranges in most of the published cases. On the other hand, mild elevations in the level of ammonia that did not cause any symptoms have been observed in patients treated with VA in whom medication did not need to be discontinued.13 Thus, we do not believe that levels of ammonia need to be monitored in all patients treated with VA, although they should be monitored in those who develop neurological symptoms.14
Several factors may be associated to the development of VHE, but the most important are polytherapy and deficiency of ornithine transcarbamylase (OTC) and of carnitine. Some anticonvulsant drugs, such as phenobarbital, phenytoin, carbamazepine, and topiramate, increase the toxicity of VA.15 OTC deficiency (an X-linked disease) is the most common inherited cause of hyperammonaemia. Male homozygotes usually die in the neonatal period, while female heterozygotes may be asymptomatic and develop hyperammonaemia secondary to VA therapy.16
Treatment of VHE consists of discontinuation of valproate therapy and control of ammonia levels using carnitine and N-carbamoylglutamate, to prevent neurotoxicity.7 NCG, a synthetic analogue of NAG, is indicated for treatment of hyperammonaemia secondary to NAGS deficit, but has been used in hyperammonaemia of other (organic acidemias, beta-oxidation disorders) or unknown aetiologies with good results.7,17 Giner and cols, report two cases in adolescents in which VA was discontinued and who were treated successfully with NCG over 24–48h,18 and recently a case of a preschooler treated for 2 months with NCG while the dose of VA was tapered off.3
We report this case due to its rarity and the difficulty of its diagnosis. We also want to emphasise that while VHE is a rare condition, it needs to be considered in the diagnosis of any neonate undergoing VA therapy with an unexplained impairment of consciousness. In our case, polytherapy could have been a contributing factor; other possible causes of hyperammonaemia were ruled out. Finally, we ought to emphasise the therapeutic efficacy of discontinuing VA therapy and administering NCG combined with other drugs in normalising ammonia levels, which resulted in full resolution of the condition without short- or medium-term sequelae.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Fernández Colomer B, Rekarte García S, García López JE, Pérez González C, Montes Granda M, Coto Cotallo GD. Encefalopatía hiperamoniémica inducida por ácido valproico en un neonato. Tratamiento con ácido carglúmico. An Pediatr. (Barc). 2014;81:251–255.
