





Despite standard clinical definitions and availability of diagnostic tests for precocious puberty, an intensive and structured investigation is needed in order to diagnose the aetiology in particular cases.
A 4-year-old, phenotypically female child was referred to paediatric endocrinology consultation for premature pubarche and thelarche. There was an acceleration of growth velocity with high levels of oestradiol and testosterone, and prepubertal FSH and LH measurements. Investigation showed bilateral gonadoblastoma as the cause of the peripheral precocious puberty.
Genetic studies revealed 46,XY karyotype with mutation c.89G>T (p.Arg30Ile) in exon 1 of the SRY gene, confirming the diagnosis of complete gonadal dysgenesis. Disorders of sexual differentiation must be considered in the approach and investigation of peripheral precocious puberty, especially in the presence of ovarian tumours, such as gonadoblastoma and dysgerminoma.
La pubertad precoz, a pesar de las definiciones clínicas estandarizadas y pruebas de diagnóstico disponibles, requiere, en ciertas situaciones una investigación exhaustiva y estructurada con el fin de conocer la causa.
Niña de 4 años de edad, fenotípicamente de sexo femenino, enviada a la consulta de endocrinología pediátrica por pubarquia y telarquia. Se observó aceleración en la tasa de crecimiento con niveles altos de estradiol y testosterona, con determinaciones prepúberes de la hormona luteinizante y foliculoestimulante. El resto del estudio de pubertad precoz periférica mostró la presencia de gonadoblastoma bilateral. El estudio genético reveló cariotipo 46 XY con mutación c.89G> T (p.Arg30Ile) en el exón 1 del gen SRY, confirmando el diagnóstico de disgenesia gonadal completa.
Los trastornos de la diferenciación sexual deben ser considerados en el abordaje y la investigación de las causas de la pubertad precoz periférica, especialmente en presencia de tumores de ovario, como gonadoblastoma y disgerminomas.
Precocious puberty (PP) is defined as the development of secondary sexual characteristics before the age of 8 years in girls and 9 years in boys.1,2
The aetiology of PP is diverse, ranging from variations of normal development, such as isolated premature thelarche, to diseases with significant comorbidity and mortality, such as germ cell tumours.
Addressing PP involves classifying it into two subtypes: central precocious puberty (CPP), which is gonadotropin-dependent (caused by early maturation of the hypothalamic–pituitary–gonadal [HPG] axis) and peripheral precocious puberty (PPP), which is gonadotropin-independent (due to excess secretion of sex hormones, androgens or oestrogens, from the adrenal glands, the gonads or exogenous sources).2
PPP may be of genetic origin (testotoxicosis; congenital adrenal hyperplasia; DAX1 gene mutation; McCune–Albright syndrome) or acquired (ovarian cyst; ovarian, testicular or adrenal tumours that produce the β subunit of human chorionic gonadotropin [β-hCG]); exogenous sex steroids).3
Acquired PPP occurs secondary to an increase in exogenous or endogenous sex steroids.
Diagnostic assessment includes an anamnesis and a detailed physical examination and additional tests, such as determination of basal and LHRH-stimulated gonadotropin levels (essential for differential diagnosis between CPP and PPP), hormone tests (testosterone, 17-β-oestradiol, dehydroepiandrosterone sulphate [DHEA-S], androstenedione and 17-hydroxyprogesterone [17-OH-progesterone], β-HCG, free thyroxine [free T4] and thyroid-stimulating hormone [TSH]) and imaging tests (hand-wrist radiograph to determine bone age, pelvic or testicular and abdominal ultrasound and cranial MRI). Finally, in the presence of strong clinical suspicion, genetic testing should be performed.3
Case historyGirl referred to paediatric endocrinology consultation at the age of 4 years 2 months with suspected precocious puberty.
She had been born to term with adequate somatometry for her gestational age and no important family, perinatal or pathological history.
She presented with appropriate psychomotor development for her age and her height and weight development. Her weight was above the 95th percentile with upward centile crossing, having ranged from the 25th to the 50th percentile up to age 2 and then crossed to values above the 95th percentile by the age of 4 (Fig. 1).
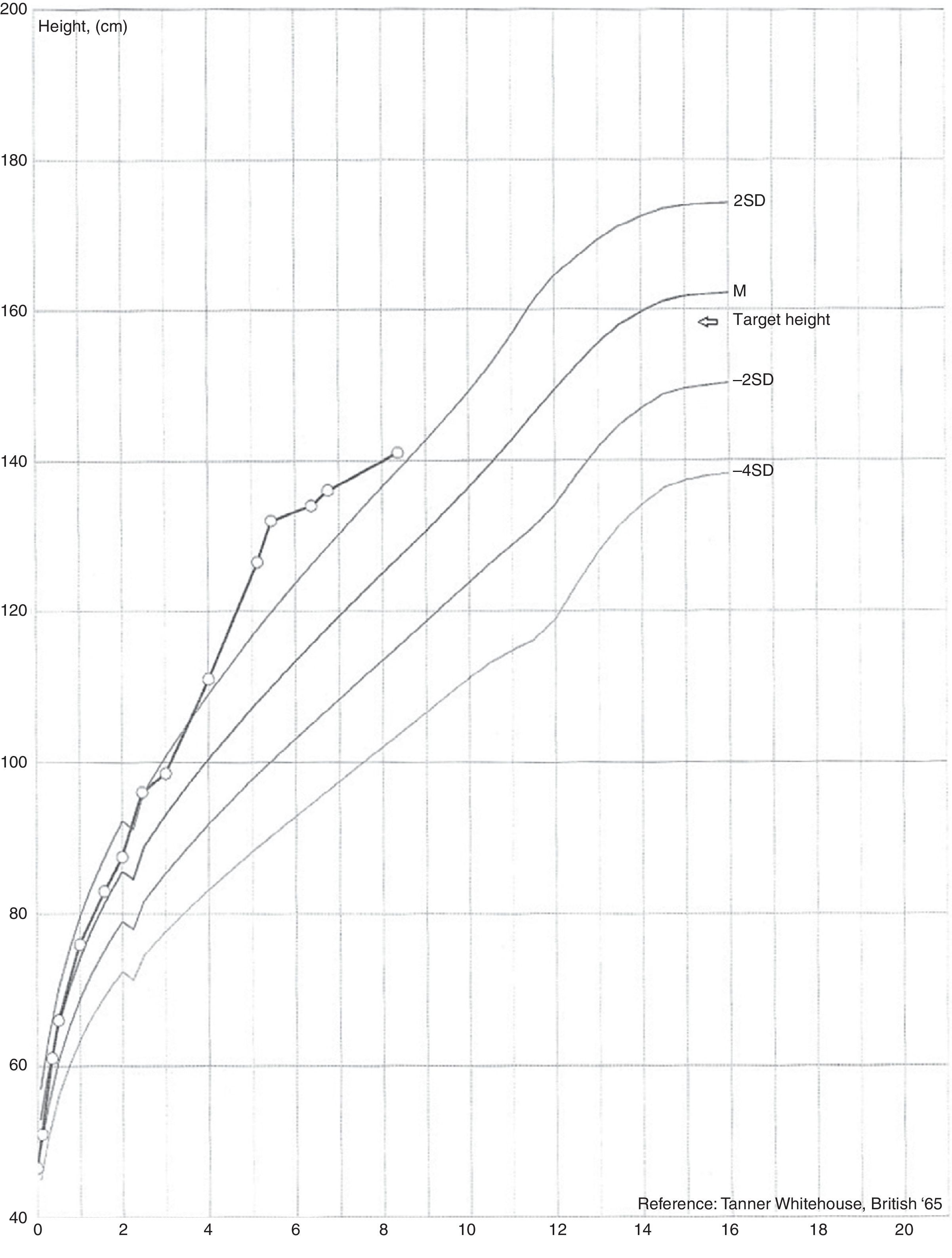
Development of secondary sexual characteristics was observed at age 4, with the appearance of pubic hair and breast budding. In her first hospital assessment her weight was 22kg (>95th percentile), height 115.5cm (standard deviation score [SDS] 3.10), growth velocity 14.81cm/year (SDS 7.13), target height 159cm (SDS −0.50) and external female genitalia with Tanner stage A1-B2-P2.
Laboratory tests revealed high levels of oestradiol, total testosterone and β-HCG (Table 1).
Hormone values and tumour markers.
| Result | Reference value | |
| 17-Hydroxyprogesterone | 90ng/dL | 4–115ng/dL |
| Dehydroepiandrosterone sulphate | 27.1μg/dL | 5–35μg/dL |
| Delta-4-androstenedione | 0.357ng/mL | 0.85–2.75ng/mL |
| Free thyroxine | 7.5μg/dL | 7.3–15μg/dL |
| Thyroid-stimulating hormone | 1.5μU/mL | 0.6–6.3μU/mL |
| IGF1 | 297ng/mL | 16–396ng/mL |
| Adrenocorticotropic hormone | 15.5pg/mL | 7–28pg/mL |
| Cortisol | 7μg/dL | 3–20μg/dL |
| Prolactin | 13.5ng/mL | 2.6–18ng/mL |
| Lactate dehydrogenase | 257U/L | 110–295U/L |
| Alpha-foetoprotein | 1.6ng/mL | 0–15ng/mL |
| β-HCG | 10.3U/L | ≤1U/L |
| FSH | 0.3mU/mL | <5mU/mL |
| LH | <0.1mU/L | <5mU/L |
| Oestradiol | 17.3pg/mL | <12.5pg/mL |
| Total testosterone | 1.21ng/mL | 0.06–0.8ng/mL |
β-HCG: beta subunit of the human chorionic gonadotropin hormone; FSH: follicle-stimulating hormone; IGF1: insulin-like growth factor 1; LH: luteinising hormone.
The levels of 17-OH-progesterone, DHEA-S, delta-4-androstenedione, free T4, TSH, adrenocorticotropic hormone, cortisol (morning level), prolactin, lactate dehydrogenase (LDH) and alpha-foetoprotein (α-FP) were normal for her age (Table 1). Serum FSH was 0.3mU/mL and LH<0.1mU/L, and the LH/RH test showed a prepubertal response (peak LH 1.1mU/mL; peak FSH 2.3mU/mL, LH/FSH ratio <1).
Bone age (BA) was 19 months in advance of chronological age (CA) (BA: 5 years 9 months, CA: 4 years 2 months). The pelvic ultrasound revealed a focal, well-circumscribed hyperechoic mass 25mm in diameter in the left adnexal region. The pelvic computed tomography revealed bilateral solid adnexal lesions, with calcified matrix, measuring 30mm on the right and 25mm on the left.
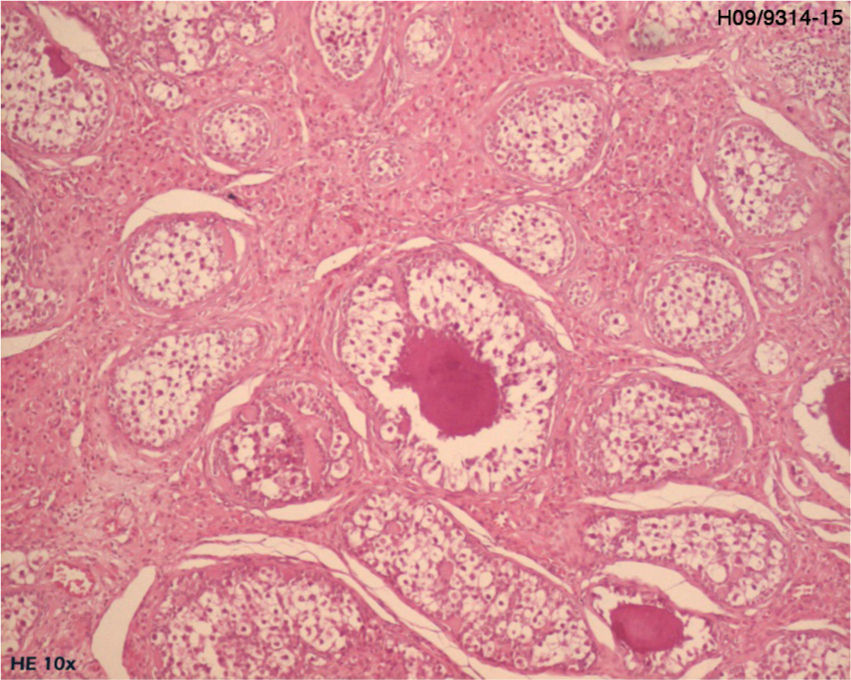
Histological examination of the biopsy specimen showed patches of tissue occupied by bilateral gonadoblastoma structures, confirmed by immunohistochemistry (CD117 and inhibin-alpha).
A laparoscopic bilateral oophorectomy was performed. Anatomohistopathological analysis confirmed the presence of bilateral gonadoblastoma (Figs. 2 and 3).

Genetic testing of peripheral blood revealed a 46,XY karyotype with mutation c.89G>T (p.Arg30Ile) in exon 1 of the SRY gene (sex-determining region of the Y chromosome), which confirmed the diagnosis of complete XY gonadal dysgenesis due to a mutation in the SRY gene.
DiscussionGonadoblastomas are rare benign tumours composed of germ cells mixed with circumscribed nests of sex cord cells, generally with a hyaline basement membrane and with diffuse or focal calcifications.4 They were first described by Scully in 1953.5
In 50% of cases there is excessive and abnormal growth of the germ cells, with progression to dysgerminoma, and occasionally other germ tumours, such as embryonic carcinoma and choriocarcinoma.6 These tumours are rarely causes of precocious puberty.7
In this case, the onset of thelarche and pubarche at age 4, with a marked acceleration of growth velocity, was indicative of an organic condition as the cause of PP. The laboratory study showed high levels of oestradiol and testosterone, with prepubertal FSH and LH levels. These facts guided the analysis of peripheral causes. The pelvic assessment identified the presence of ovarian masses which, in conjunction with the high β-HCG level, suggested a tumoural condition as the diagnosis.
β-HCG is a glycoprotein hormone, which, as well as being associated with pregnancy, is considered a tumour marker for gynaecological cancer, and also, more rarely, for non-gynaecological cancer.8 The tumours are hormonally active, with the ability to produce β-HCG.9
Other possible germ cell tumour markers, such as LDH and α-FP, were within normal values. Capito et al., in a study carried out with 11 patients with 46,XY pure gonadal dysgenesis, also described the presence of gonadoblastoma and dysgerminoma with normal α-FP levels in a 17-year-old patient with incomplete pubertal development for her age.10
In the case presented here, the biopsy confirmed a tumoural condition, which was identified as bilateral gonadoblastoma.
These tumours occur almost exclusively in patients with gonadal dysgenesis associated with the presence of a Y chromosome, the most frequent karyotypes in these situations being 46,XY and 45,X/46,XY mosaicism.6
Clinical presentation is variable, ranging from the absence of symptoms to different degrees of virilisation or feminisation and the presence of abdominal mass.11
Presentation with signs of precocious puberty seems to be related to autonomous sex steroid secretion, which occurs in 15% of cases, being conditioned by tumour components derived from sex cord cells. Androgen secretion induces virilisation, or isosexual pseudopuberty, if oestrogen secretion dominates.10
In this case, this strong association of gonadoblastomas with disorders of sexual differentiation led us to carry out a genetic study, which confirmed the diagnosis of 46,XY complete gonadal dysgenesis with the identification of mutation c.89G>T (p.Arg30Ile) in exon 1 of the SRY gene. This mutation was first described as a cause of gonadal dysgenesis by Assumpção et al. in 2002.12
Complete gonadal dysgenesis is characterised by gonadal streaks, normal development of the Müller ducts and external female genitalia.10,13 This disruption of sexual differentiation is the result of a failure of foetal testicular development, secondary to mutations in certain genes, such as SRY, SOX9, SF1, DAX1 and WT1.10,14 Mutations of the SRY gene represent only 10–20% of 46,XY disorders of sexual differentiation, which is due in the remaining cases to mutations in other genes among those mentioned.13
Oophorectomy remains the treatment of choice due to the high risk of malignant transformation.6
As well as oophorectomy, the treatment includes hormone therapy and fertility induction, because of the infertility of all these patients.
In conclusion, we wish to emphasise how important it is for health care professionals to be familiar with the physiology of normal puberty, so as to be able to recognise anomalies in pubertal development, whose aetiology may entail high morbidity and mortality, as in the case of ovarian tumours.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Santalha M, Amaral B, Pereira J, Ribeiro L, João Oliveira M, Figueiredo S, et al. Pubertad precoz periférica: disgenesia gonadal completa 46 XY. An Pediatr (Barc). 2014;81:246–250.
