




El seudohipoparatiroidismo (PHP) es una entidad rara, caracterizada por resistencia tisular a la hormona paratiroidea (PTH). Los 2 subtipos principales, PHP-Ia y PHP-Ib, son causados por alteraciones en el gen GNAS (20q13.3), que codifica para la proteína Gsα, esencial para la acción de la PTH y otras hormonas.
El PHP-Ia se asocia a diversas alteraciones hormonales, osteodistrofia hereditaria de Albright (AHO) y actividad reducida de Gsα. Está causado por mutaciones inactivantes del gen GNAS. El PHP-Ib presenta resistencia aislada a la PTH, sin AHO y con actividad Gsα normal o levemente baja. Se asocia a defectos en la impronta de GNAS.
Se presentan 2 casos con PHP-Ia y PHP-Ib, ahondando en su clínica y en el diagnóstico diferencial frente a afecciones similares.
Pseudohypoparathyroidism (PHP) is a rare disorder, characterized by a tissue resistance to parathyroid hormone (PTH). The two main subtypes of PHP, PHPIa and PHPIb, are caused by alterations in the GNAS locus (20q13.3), which encodes the Gsα protein, essential for the action of PTH and other hormones.
PHP-Ia is associated with several hormone resistances, Albright hereditary osteodystrophy (AHO), and reduced Gsα activity. It is caused by inactivating mutations in the GNAS gene. PHP-Ib presents with isolated resistance to PTH, without AHO and with normal to low Gsα activity. It is related to imprinting defects in GNAS.
Two unrelated cases of PHP-Ia and PHP-Ib are presented here, focusing on their clinical aspects and in the differential diagnosis with similar pathologies.
Muchas hormonas peptídicas, como la hormona paratiroidea (PTH), la hormona estimuladora de tiroides (TSH), las gonadotropinas (LH y FSH), estimuladora de la hormona de crecimiento, etc., así como neurotransmisores y factores autocrinos y paracrinos, ejercen sus acciones a través de receptores acoplados a la proteína de membrana Gsα1,2 que, tras ser activada por el complejo hormona-receptor, desencadena procesos de señalización celular a través de un segundo mensajero, el AMP cíclico (fig. 1a). Esta cascada de señales incluye fosforilaciones mediadas por la proteincinasa A (PKA), mientras que las fosfodiesterasas actúan como reguladores negativas de la vía (fig. 1b)3. Así, alteraciones en los receptores específicos provocan resistencias selectivas a dichas hormonas, mientras que alteraciones de la vía AMPc/PKA afectan simultáneamente a diversos sistemas metabólicos.
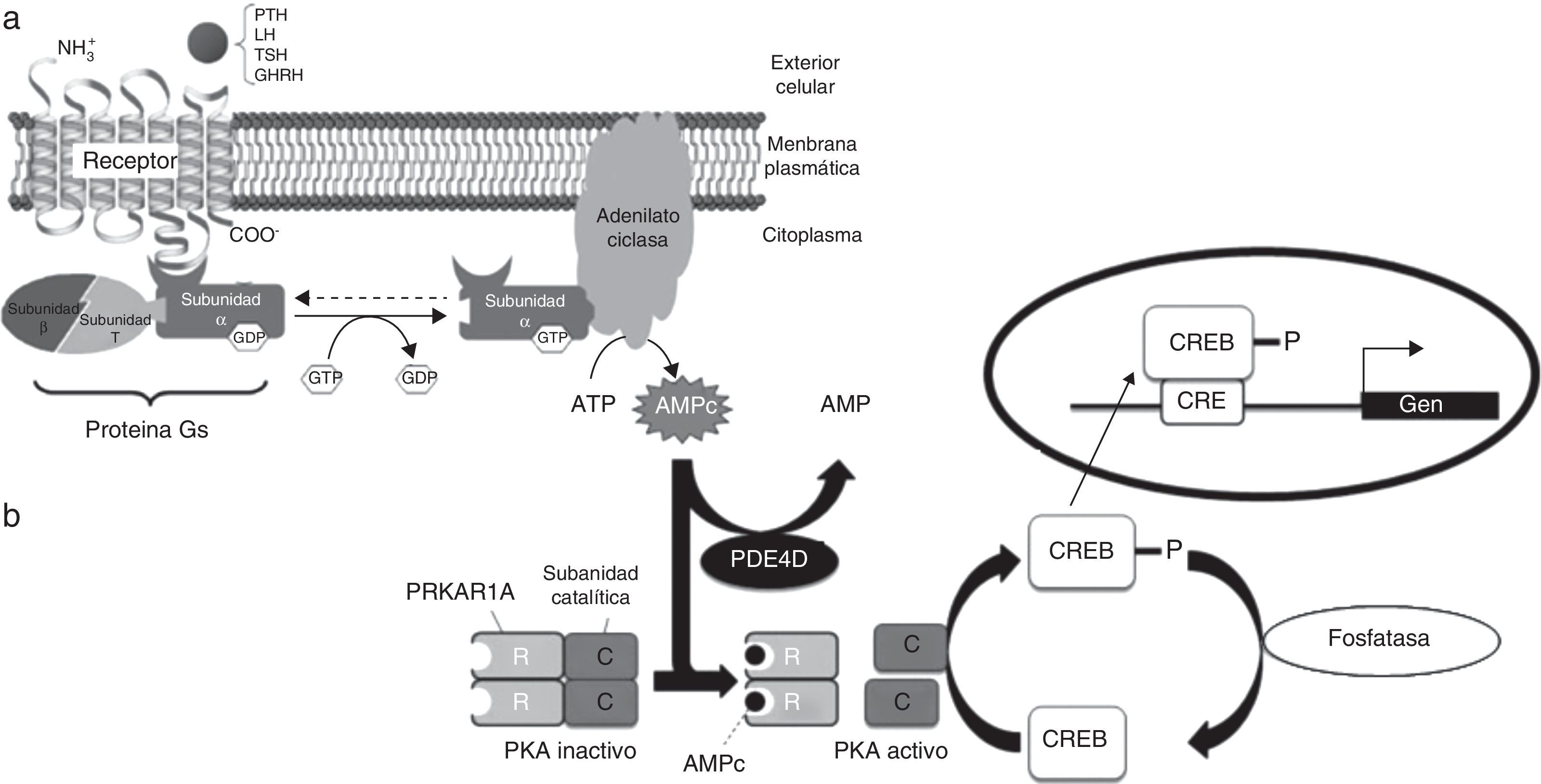
Esquema de la vía del AMPc/proteincinasa A. a) Mecanismo de acción de las hormonas cuyo receptor está acoplado a proteína Gsα. Al unirse la hormona al receptor, se activa la subunidad α de la proteína Gs. Esta interacciona con la adenilato ciclasa, tras lo que se produce la síntesis de AMPc, que ejerce su función como segundo mensajero. Alteraciones a nivel de GNAS (gen que codifica para Gsα) dan lugar a PHP, PPHP, AHO, etc. b) La unión del AMPc a PRKAR1A, la subunidad reguladora dependiente de AMPc, lleva a la disociación y activación de la proteincinasa A. CREB (proteína de unión a los elementos de respuesta al AMPc) se fosforila, con lo que se transloca al núcleo y modifica la expresión de los genes «aguas abajo» de la ruta. La actividad de la fosfodiesterasa PDE4D modula los niveles de AMPc. Alteraciones a nivel de PDE4D y PRKAR1A dan lugar a acrodisostosis, entidad clínicamente relacionada con la AHO.
El seudohipoparatiroidismo (PHP) es un grupo de endocrinopatías raras, caracterizadas por hipocalcemia, hiperfosfatemia y aumento de la hormona paratiroidea (PTH), debido a una resistencia variable a dicha hormona en sus órganos diana, fundamentalmente el túbulo renal proximal2,4,5. El diagnóstico de PHP se realiza, casi siempre, en la infancia, por hipocalcemia o por un peculiar fenotipo llamado osteodistrofia hereditaria de Albright (AHO)4, caracterizado por talla baja, obesidad, cara redondeada, braquidactilia, calcificaciones ectópicas y/o retraso mental. En función del fenotipo y la bioquímica asociada, el PHP se divide en diversos subtipos, siendo los 2 más importantes el PHP-Ia (OMIM #103580) y el PHP-Ib (OMIM #603233).
Pacientes con PHP-Ia presentan múltiple resistencia hormonal (PTH, TSH, gonadotropinas), fenotipo AHO y reducción en la actividad de Gsα del 50%. Los pacientes con PHP-Ib presentan resistencia a la PTH sin AHO, con actividad de Gsα habitualmente normal. Estas formas de PHP están causadas por defectos en el locus GNAS (20q13.2-13.3), donde se encuentra el gen GNAS, codificante para la proteína Gsα6,7.
En este trabajo se presenta el estudio clínico y molecular de 2 casos con PHP-Ia y PHP-Ib.
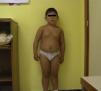
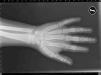
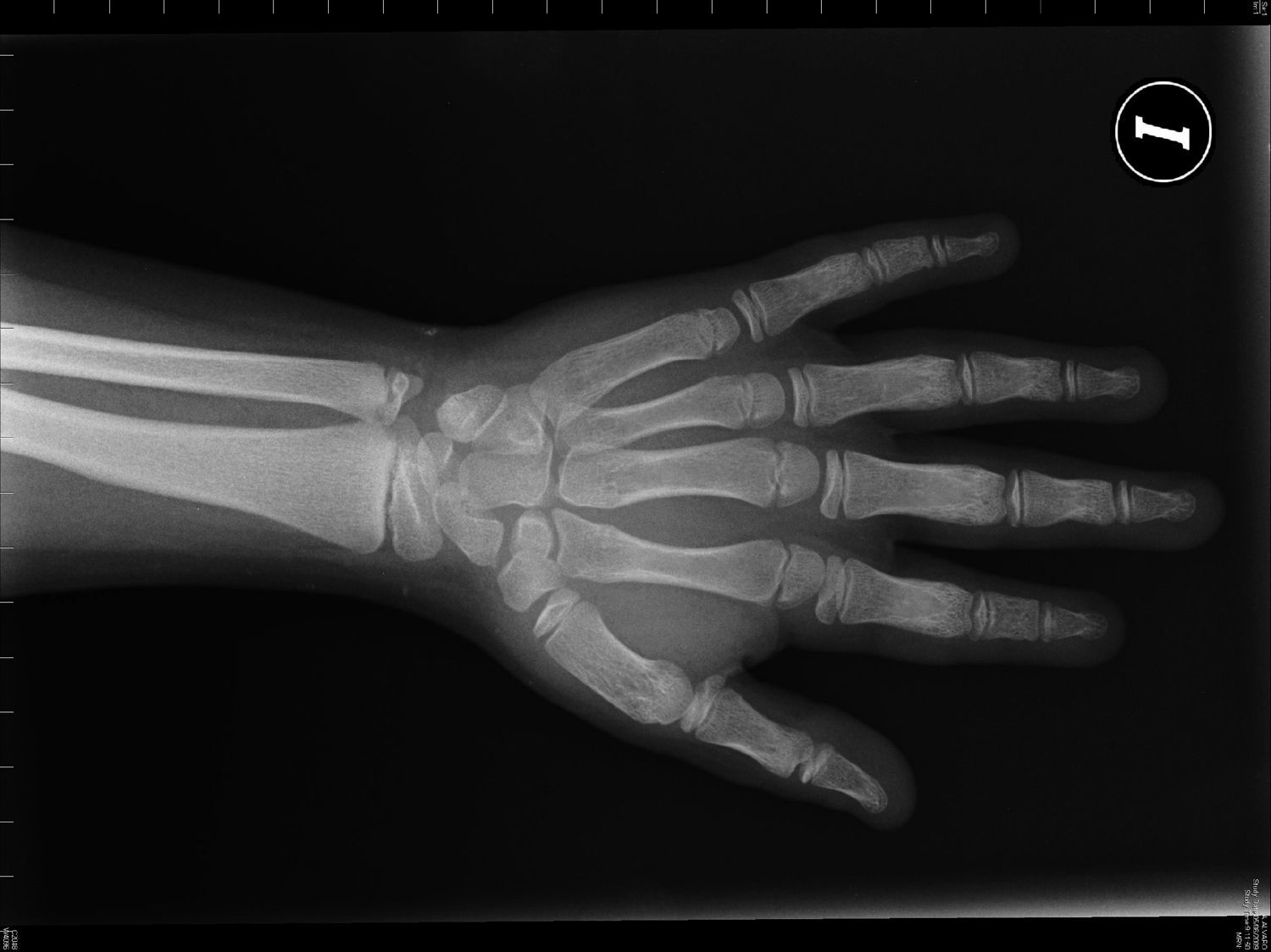
Pacientes y métodosCaso 1Varón de 9 años, hijo de padres sanos, no consanguíneos. Presentó hiperbilirrubinemia en el periodo neonatal. Estudiado por primera vez a los 2 años de edad por calcificaciones subcutáneas en antebrazos, abdomen, muslos y piernas. La exploración física reveló obesidad, cara redonda, cuello corto y manos pequeñas (fig. 2). Sin desarrollo puberal, peso 20kg (DE 3,2), talla 90cm (DE –1,1), índice de masa corporal (IMC) 24,7 (DE 6,9), lesiones máculo-papulosas induradas en el tronco y las extremidades. Bioquímica en sangre: calcio (Ca) total 9,3mg/dl (rango normal 8,8-10,8); Ca iónico 4,9mg/dl (4,9-5,5); fósforo (P) 5,6mg/dl (2,9-5,1); PTH intacta 830 pg/ml (0-68,2); tiroxina T4 libre (T4L) 0,9 ng/dl (0,7-2,0) y TSH 2,9μU/ml (0,49-4,67). Tomografía computarizada cerebral (TCC) normal. A los 8 años de edad presenta disminución de los niveles de Ca total (8,1mg/dl) y Ca iónico (4,36mg/dl) y aumento de niveles de P (6,7mg/dl) y PTH (1.716 pg/ml); el calcitriol 56 pg/ml (normal 16-56) y la T4L (0,8 ng/dl) fueron normales, y la TSH fue elevada (9,7μU/ml). La TCC muestra pequeñas calcificaciones en plexos coroideos. No han aparecido nuevas calcificaciones subcutáneas. En la radiografía de mano izquierda se observa braquidactilia, osteoporosis y aceleración de la edad ósea (edad ósea, 12 años; edad cronológica, 9 años) (fig. 3). Se realiza diagnóstico clínico de PHP-Ia e hipotiroidismo e inicia tratamiento con L-tiroxina, calcio y calcitriol, con normalización de las cifras de Ca, P, T4L y TSH, mientras que la PTH persiste elevada.
Caso 2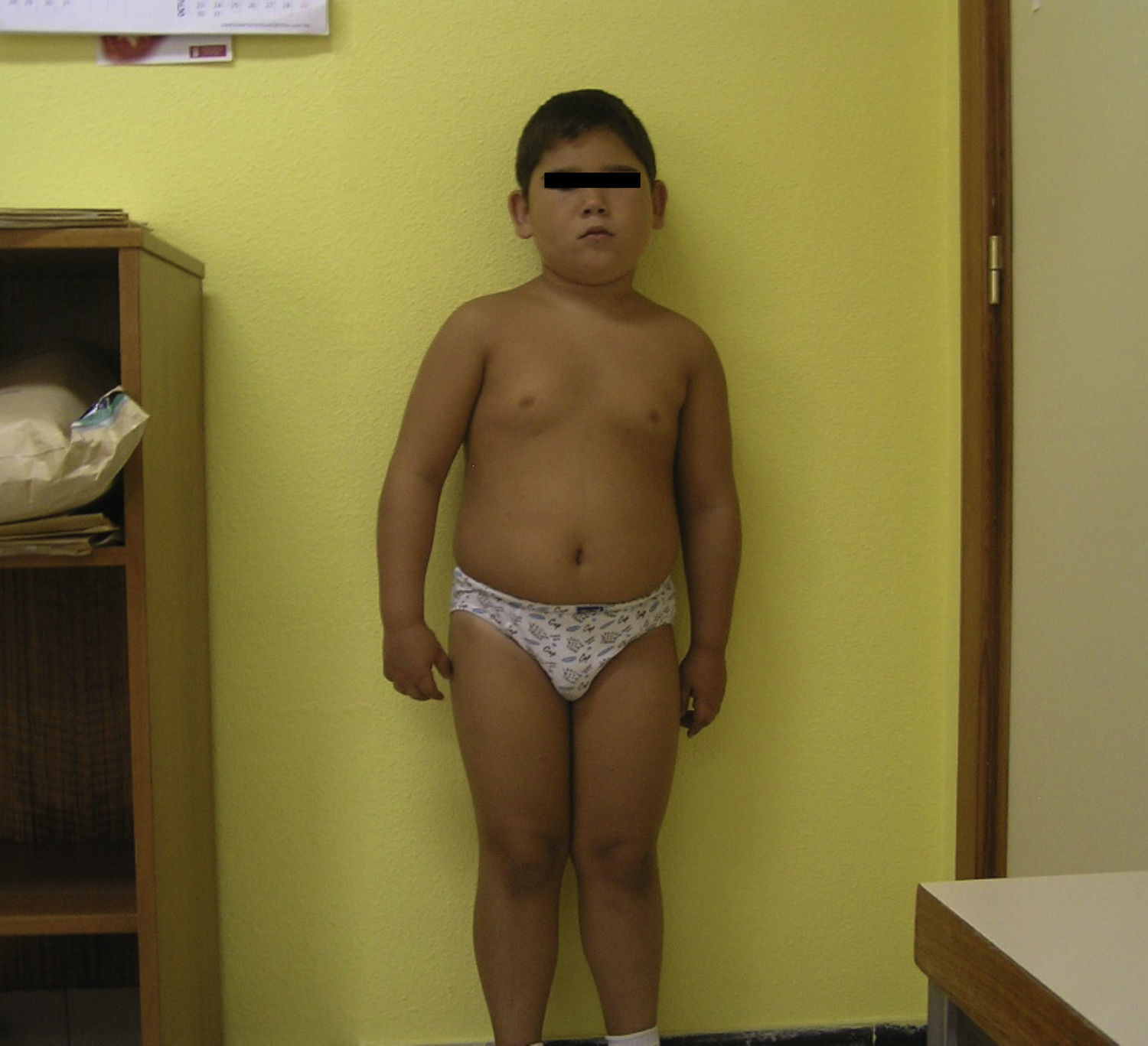
Varón de 11 años, sin antecedentes familiares de interés, remitido a endocrinología por hipocalcemia severa detectada por cuadro de mioclonías y espasmos de miembros inferiores (MMII), empeorado en las 2 semanas previas. Refería desde los 3 años tratamiento rehabilitador por torpeza motora, hipertonía de MMII y dislalia. La exploración clínica muestra peso 53,2kg (DE 1,18), talla 150cm (DE 0,79) e IMC 23,6 (DE 1,65), leve hipertonía de MMII, retracción aquílea, marcha con rigidez, torpeza motora y pies en garra, sin desarrollo puberal y ausencia de fenotipo peculiar. En la analítica destacaban: Ca total 5,3mg/dl; Ca iónico 2,34mg/dl; P 10,6mg/dl; PTH intacta 674 pg/ml; TSH 3,38 mU/l y T4L 1,0 ng/dl; calcitriol normal. La TCC muestra múltiples calcificaciones bilaterales y simétricas en ganglios basales, con calcificaciones corticosubcorticales en parénquima de lóbulos frontales, temporales y parietales. Con estos hallazgos, se realizó el diagnóstico de PHP-Ib. Se inició tratamiento con calcio y calcitriol, con desaparición de los síntomas y normalización de las cifras de Ca, P y PTH a los 12 meses de tratamiento.
No fue posible realizar en ninguno de los 2 casos el test de Ellsworth-Howard, ni la determinación de la actividad eritrocitaria de la proteína Gsα.
Análisis molecularEl ADN fue extraído de sangre total usando el QIAamp DNABlood Minikit (Qiagen, Alemania). Los 13 exones codificantes del gen GNAS se estudiaron por reacción en cadena de la polimerasa, purificación con Exo-Sap (USB, EE. UU.) y secuenciación directa en un secuenciador ABI3130xl (Applied Biosystems, EE. UU.). Las secuencias fueron analizadas y comparadas con la referencia del gen GNAS (Ensembl ENST00000371085) usando los software Sequencing Analysis v5.2 y SeqScape v2.5 (Applied Biosystems).
Se usó la técnica de methylation-specific multiplex-ligation-dependent probe amplification (MS-MLPA) para estudiar posibles deleciones, duplicaciones y la metilación del locus GNAS, mediante el kit ME031 (MRC-Holland, Holanda) en un secuenciador ABI3130xl, usando el software GeneMapper v4.0 (Applied Biosystems).
Resultados molecularesEl caso 1 (PHP-Ia) presentó una mutación en el exón 6 del gen GNAS: una duplicación en heterocigosis de 2 nucleótidos, con alteración de la pauta de lectura a partir del aminoácido 171 y aparición de un codón de parada prematuro (p.Leu171Serfs*2) (fig. 4a). Ninguno de sus progenitores presentaba dicha alteración.
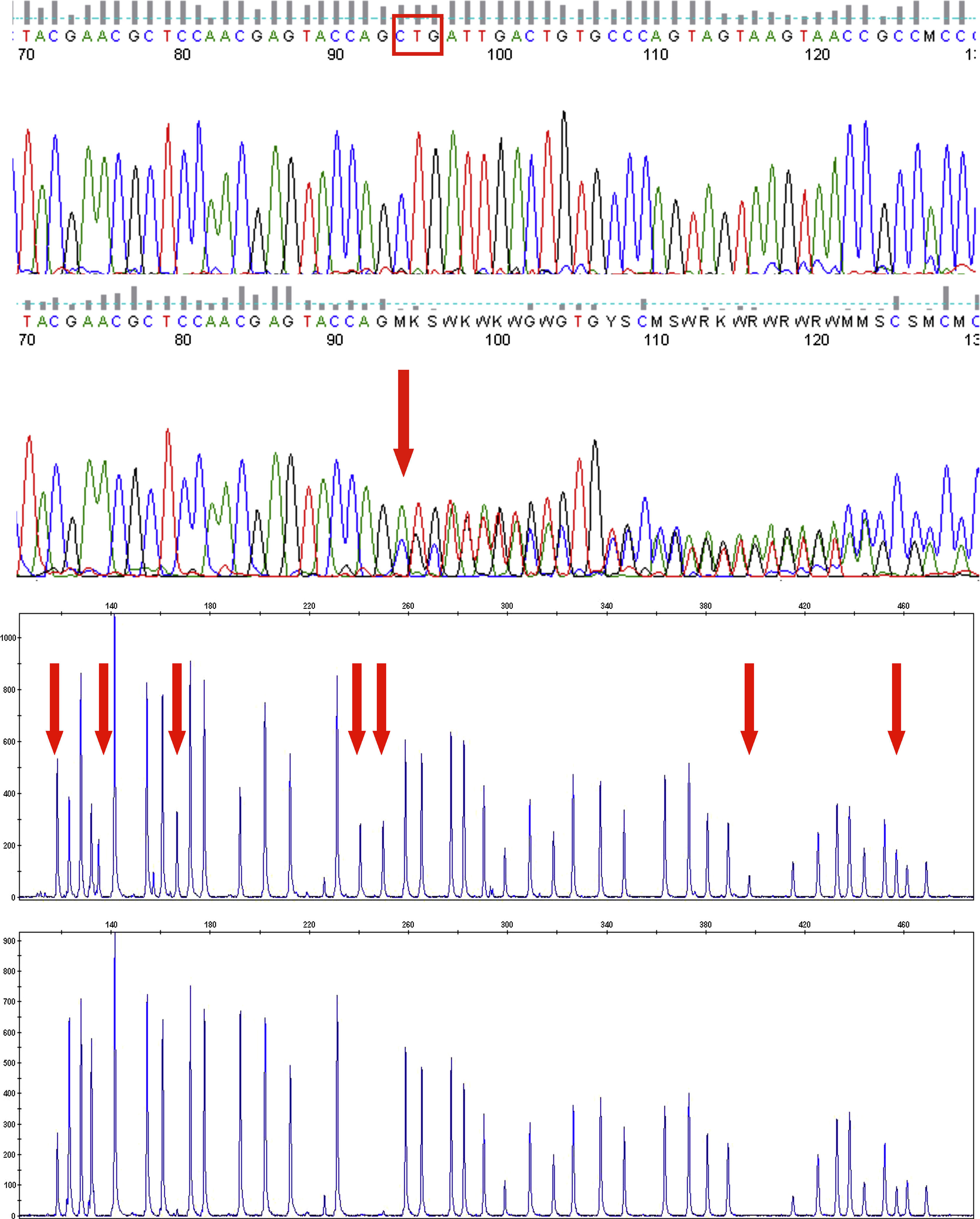
a) Electroferograma de la región del exón 6 del gen GNAS, para un control (panel superior) y el caso 1 (panel inferior). La flecha indica el lugar donde se produce la inserción en heterocigosis de 2 nucleótidos, que conlleva a nivel proteico la mutación p.Leu171Serfs*2. El recuadro en el panel superior indica los nucleótidos que codifican para el aminoácido leucina 171. b) Electroferograma que muestra el análisis por MS-MLPA del locus GNAS, para un control (panel superior) y el caso 2 (panel inferior). Las flechas indican las sondas que se pierden de manera total o parcial. Tras el análisis cuantitativo se demuestra que el paciente presenta pérdida parcial de metilación en XLαs y completa para el exón A/B.
El caso 2 (PHP-Ib) presenta pérdida parcial de metilación en XLαs y completa para el exón A/B (fig. 4b). El estudio fue negativo para sus padres y hermana.
DiscusiónEl locus GNAS está sometido a impronta, por lo que la expresión fenotípica de las alteraciones moleculares varía en función de su origen parental, así como del tejido donde se producen8-10. Los estudios moleculares han permitido caracterizar genéticamente los diferentes subtipos de PHP: el PHP-Ia se debe a mutaciones inactivantes en el alelo materno del gen GNAS, que codifica la proteína Gsα2,10-12. Clínicamente, se caracteriza por fenotipo AHO4 y resistencia hormonal, que cursa con una reducción de la actividad hormonal dependiente de la proteína Gsα (PTH, TSH, gonadotropinas, glucagón y somatotropina). La actividad de Gsα está reducida al 50%. El PHP-Ib se caracteriza por resistencia a la PTH, ausencia de fenotipo AHO y de resistencia plurihormonal2,6 (a veces hay moderada resistencia a TSH). La actividad de Gsα es normal o levemente baja. Molecularmente, el PHP-Ib se debe a cambios del patrón de metilación del locus GNAS13-15 (pérdida de metilación del exón A/B, a veces combinado con defectos epigenéticos de otras regiones del locus GNAS).
El caso 1 de este trabajo presenta PHP e hipotiroidismo, expresión de una resistencia hormonal variable en los tejidos periféricos, frecuente en el PHP-Ia. Además, la AHO orienta el diagnóstico hacia un PHP-Ia. En el caso 2, la ausencia de AHO y de resistencia plurihormonal dirige el diagnóstico hacia el PHP-Ib. En ambos casos, los resultados moleculares son los típicamente asociados: en el PHP-Ia, una mutación en heterocigosis en GNAS10-12 y en el PHP-Ib, alteraciones en el patrón de metilación del locus GNAS13-15.
Trabajos recientes han descrito defectos de metilación en algunos pacientes con PHP y fenotipo AHO8,9,14,16, solapamiento también referido en la actividad eritrocitaria de la proteína Gsα17, indicando una posible superposición molecular y clínica entre los 2 subtipos18. Además, alteraciones de la vía AMPc/PKA a diferentes niveles (fig. 1) son responsables de una serie de síndromes endocrinos con fenotipos en ocasiones similares, aunque con ligeras diferencias19. Así, la acrodisostosis (OMIM #101800), que cursa con disostosis facial, estatura baja, braquidactilia severa, en ocasiones resistencia plurihormonal (PTH y tirotropina), posible retraso mental, etc., se asemeja a la AHO, aunque está causada por alteraciones en los genes PPKAR1A y PDE4D. De ahí la importancia del análisis molecular del locus GNAS en los pacientes con sospecha de PHP para establecer un diagnóstico diferencial frente a otras entidades de la misma vía metabólica, con fenotipos similares pero cuyas causas genéticas subyacen en otros genes, y poder realizar así un adecuado consejo clínico y genético20. La ausencia de alteraciones en el locus GNAS en pacientes con fenotipo similar al fenotipo AHO y/o resistencia plurihormonal debe dirigir el estudio hacia PPKAR1A y PDE4D.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
