


Pachyonychia congenita ([PC], OMIM #167200 and #167210) is a rare genodermatosis, with 1000 cases in patients from 270 families described to date. It has a pattern of autosomal dominant inheritance (40% de novo mutations), with complete penetrance and a variable expression.1 Its historical classification includes two types: type 1 (PC-1 or Jadassohn-Lewandowsky syndrome) and type 2 (PC-2 or Jackson-Lawler syndrome).1–3 At present, it is classified based on the gene involved in keratin synthesis where the mutation is found: PC-K6a (caused by mutations in the KRT6A gene, identified in 44% of the families), PC-K6b (KRT6B, 5% of families), PC-K6c (KRT6C, 2% of families) at the 12q13.13 locus, PC-K16 (KRT16, 25% of families) and PC-K17 (KRT17, remaining 24% of families) at the 17q21.2 locus.1,3 Mutations in the KRT6A and KRT16 genes correspond to the clinical features of PC-1; while mutations in the KRT6B and KRT17 correspond to PC-2.
We present the case of a boy that received a diagnosis of PC associated with a mutation in the KRT17 gene at age 14 months.
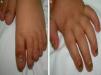
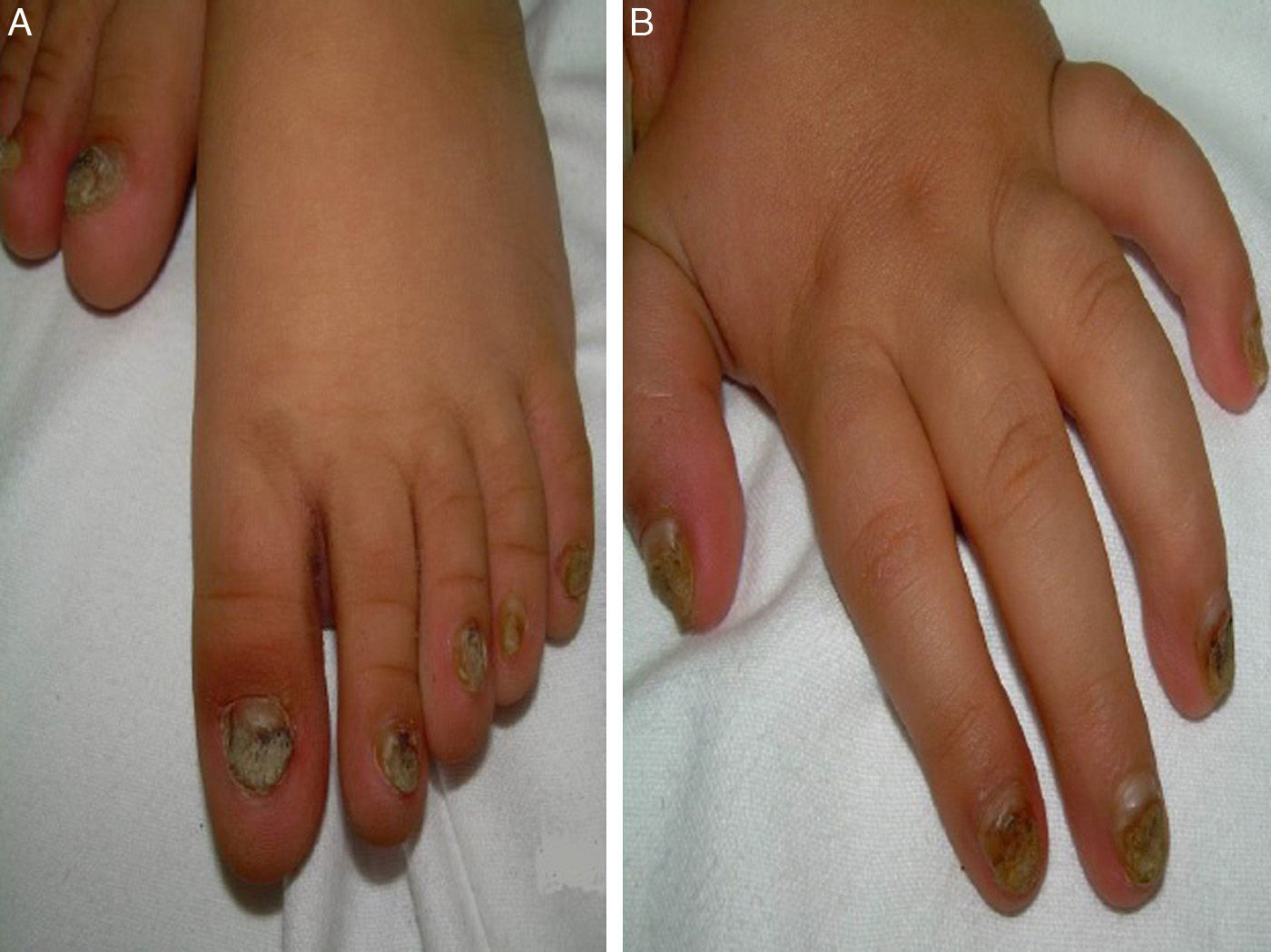
A 14-month-old boy of Moroccan descent visited the emergency department due to a respiratory infection. The physical examination revealed thickened and deformed nails in hands and feet, distal subungual hyperkeratosis and chromonychia (Fig. 1). He had been admitted at birth due to the presence of two natal teeth in the lower gum that caused bleeding and difficulty feeding, requiring extraction. The relevant family history consisted of natal teeth in one uncle and one aunt (both paternal) in the absence of ungual involvement.
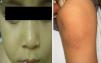
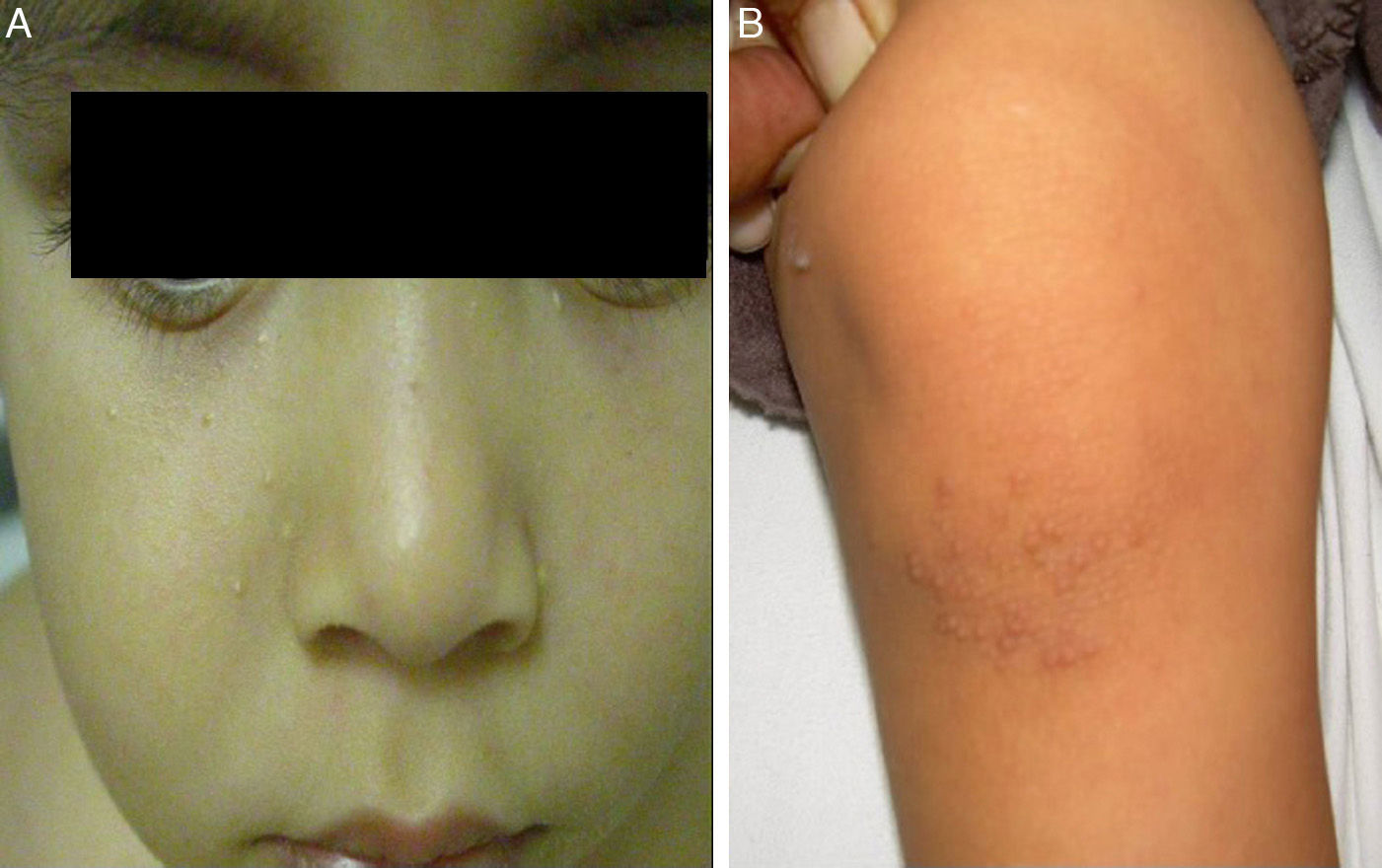
The initial differential diagnosis included congenital nail candidiasis and PC. A nail culture was performed that tested positive for Candida albicans (C. albicans), with subsequent results being negative. The disease progressed with the development of steatocystomas in the cheeks and trunk and keratosis pilaris in the knees (Fig. 2) as well as painless plantar calluses, with no palmar keratoderma. Given the clinical suspicion of PC, sequencing of the KRT17 gene was requested, which detected a heterozygous missense point mutation (c.284T>C; p.Leu95Pro).
The results of genetic testing of the parents were negative, suggesting a de novo mutation. The patient is currently 4 years of age and undergoing routine treatment with emollients and periodical nail plate grinding.
The main sign of PC is nail dystrophy, most commonly involving the two distal thirds of the nail plate. It may be associated with nail brittleness, chronic perionyxis and a predisposition to infection by C. albicans or bacterial pathogens. Therefore, the positive test result for Candida did not rule out a diagnosis of PC in our patient. Natal teeth are usually the earliest clinical manifestation. Palmoplantar hyperkeratosis is the most frequent manifestation, and is usually associated with hyperhidrosis and blisters. Other signs include follicular keratosis in arms, legs and buttocks and oral leukokeratosis. When the latter involves the larynx it causes hoarseness, and if it involves the eardrum it may cause deafness. Steatocystoma multiplex located in the trunk, limbs and underarms develops during adolescence.2 There is no cure for the disease. Painful palmoplantar keratoderma is usually the most pressing complaint. Treatment with topical keratolytic agents or retinoids may offer temporary relief, while oral retinoids are prescribed for more severe cases. Nail involvement requires treatment with emollients, grinding of the nail plate and occasionally ablation. Steatocystoma multiplex and pilosebaceous cysts can be treated with puncture and drainage of their contents.2
The heterozygous mutation c.284T>C (p.Leu95Pro) in gene KRT17 has been reported previously in another patient with PC4 and in a case recorded in the International Pachyonychia Congenita Research Registry (IPCRR).3 The former was a sporadic case that did not present with natal teeth, and the latter a familial case with the same phenotype as our patient's. There is some degree of correlation between phenotype and genotype in both types of PC. Cases of PC-1 present more frequently with oral leukokeratosis (88% of KRT6A mutations, 41% of KRT16 mutations), while natal teeth and steatocystomas are more characteristic of PC-2 (76% and 67%, respectively, in KRT17 mutations).1 This must be taken into account in the periodic evaluation of these patients. The prevalence of natal teeth in isolation is of one case in every 2000–3000 live births,5 which is how natal teeth were interpreted in the paternal aunt and uncle of the patient.
Molecular characterisation of PC allows for appropriate genetic counselling. Our patient has a 50% risk of transmitting PC to his offspring, and detection of the causal mutation will allow prenatal or preimplantation diagnosis in the future. As this was a de novo case, the risk of recurrence in future siblings is estimated at a maximum of 1% due to the possibility of germline mosaicism in one of the parents.6
Despite the presence of key phenotypical features in the first year of life, only 25% of children receive the diagnosis during this period, which leads to inadequate management and advice in most cases.7
We want to thank the Pachyonychia Congenita Project of the International Pachyonychia Congenita Research Registry (IPCRR-PC Project) of Salt Lake City, Utah, United States, for the inclusion of the patient for molecular testing by F.J.D. Smith and W.H.I. McLean of the University of Dundee in the United Kingdom.We also thank the family of the patient for their collaboration and for consenting to the publication of his clinical data and photographs.
Please cite this article as: Micol-Martínez O, López-González V, Garcia-Marcos PW, Martínez-Menchón T, Guillén-Navarro E. Paquioniquia congénita: nuevo caso asociado al gen KRT17. An Pediatr (Barc). 2016;84:174–176.
