


La paquioniquia congénita ([PC], OMIM #167200 y #167210) es una genodermatosis rara, Hay 1.000 pacientes descritos de 270 familias diferentes hasta la fecha. La PC presenta un patrón de herencia autosómica dominante (40% mutaciones de novo), de penetrancia completa y expresividad variable1. La clasificación clínica clásica incluye 2 subtipos: subtipo 1 (PC-1 o síndrome Jadassohn-Lewandowski) y subtipo 2 (PC-2 o síndrome Jackson-Lawler)1–3. En la actualidad, la clasificación se realiza en función del gen mutado implicado en la síntesis de queratina: PC-K6a (causada por mutaciones en KRT6A, identificadas en el 44% de las familias), PC-K6b (KRT6B, en el 5% de las familias), PC-K6c (KRT6C, en el 2% de las familias) localizados en 12q13.13, PC-K16 (KRT16, en el 25% de las familias) y PC-K17 (KRT17, en el 24% restante) en 17q21.21,3. Las mutaciones localizadas en KRT6A y KRT16 corresponderían clínicamente con PC-1; mientras las localizadas en KRT6B y KRT17 corresponden con PC-2.
Presentamos el caso de un niño diagnosticado de PC a los 14 meses de vida asociado a una mutación en el gen KRT17.
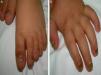
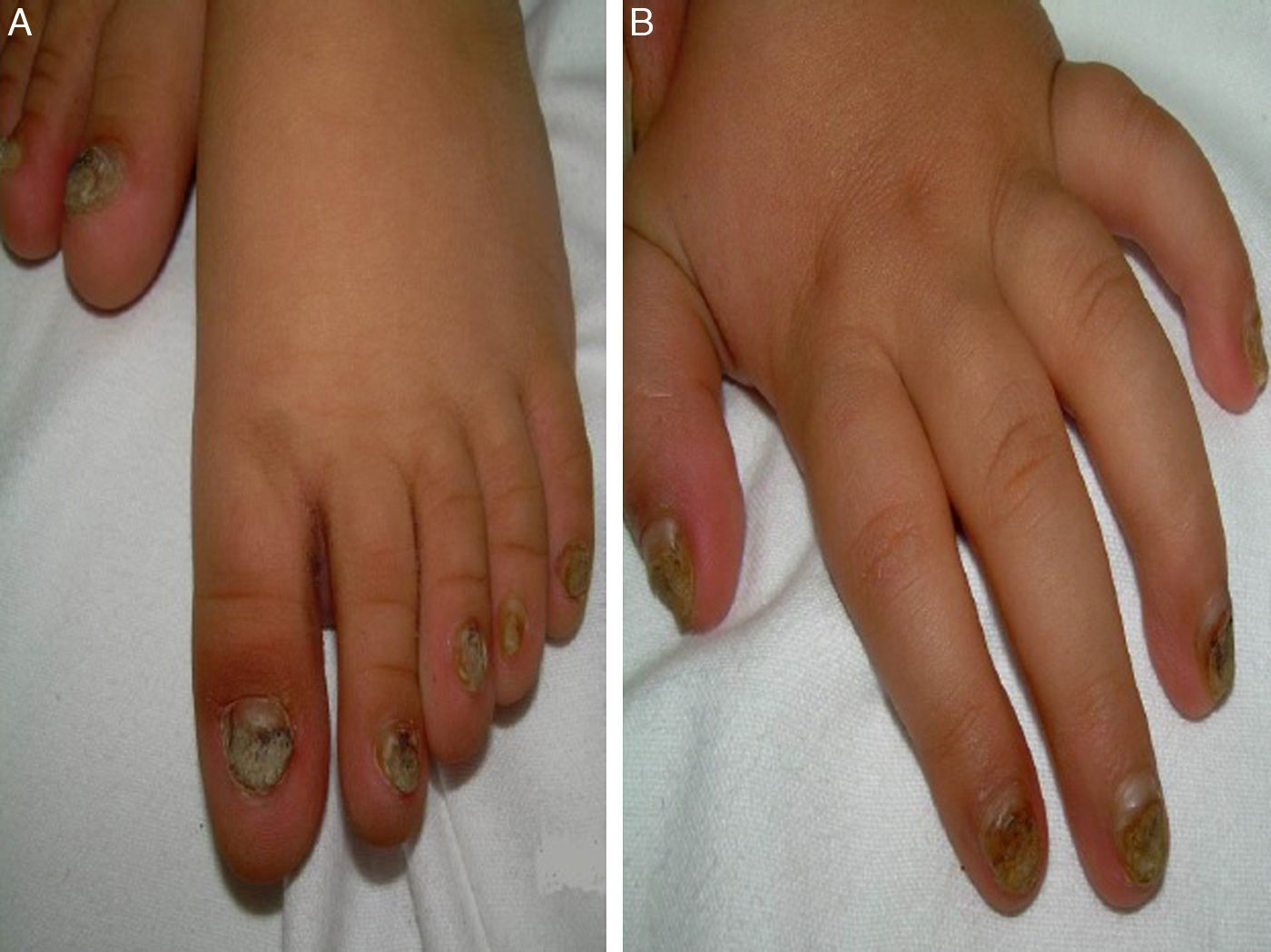
Niño de 14 meses, de origen marroquí, acude a urgencias por infección respiratoria, detectando a la exploración engrosamiento y deformidad ungueal en manos y pies, hiperqueratosis subungueal distal y cromoniquia (fig. 1). Ingresó al nacimiento por presencia de 2 piezas dentarias congénitas inferiores que ocasionaron sangrado y dificultad para la alimentación, precisando exodoncia. Entre los antecedentes familiares destacaba que su tío y tía paternos presentaron dientes natales, sin afectación ungueal asociada.
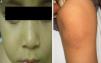
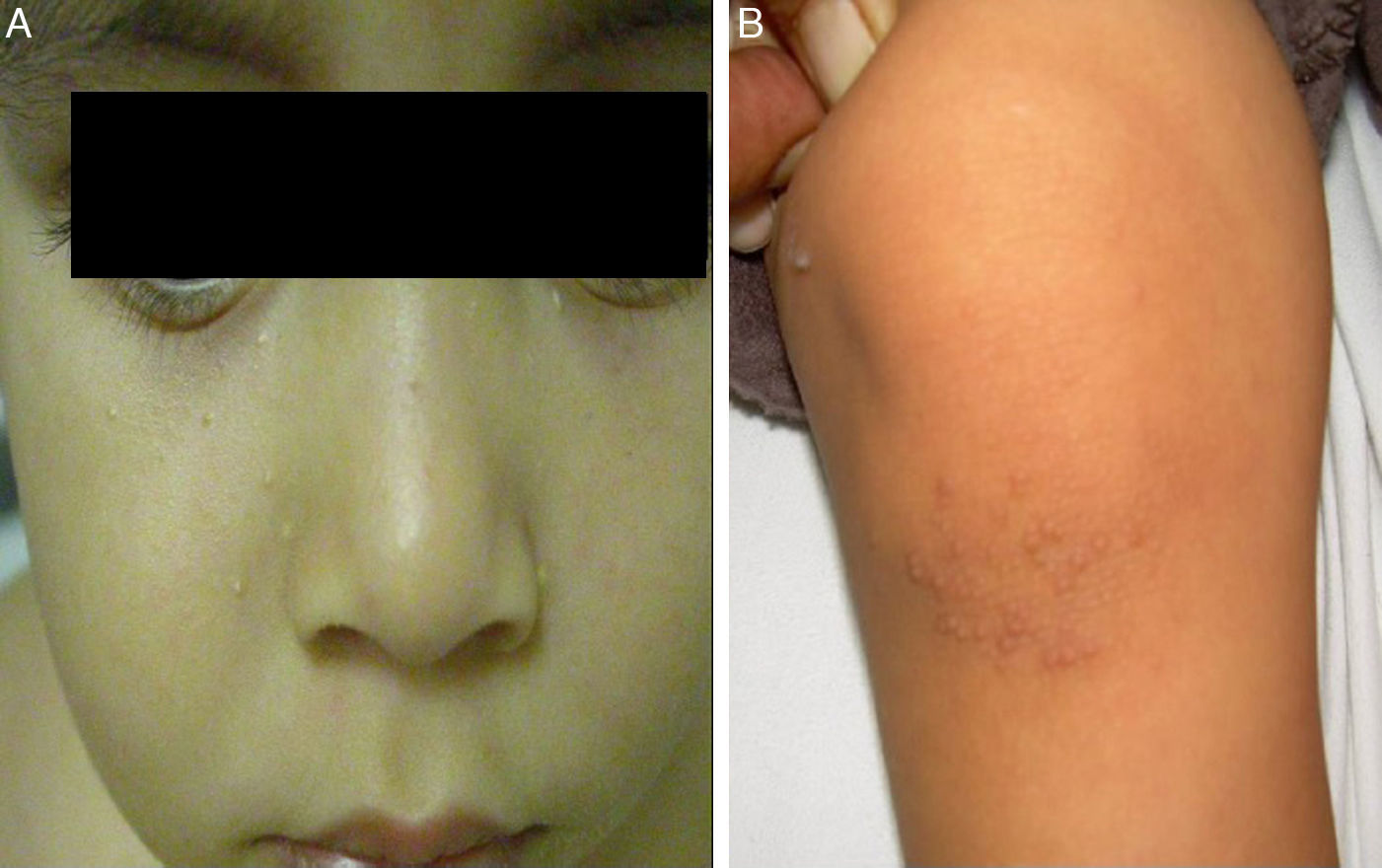
Inicialmente se planteó el diagnóstico diferencial entre candidiasis congénita ungueal y PC. Se recogió cultivo ungueal resultando positivo para Candida albicans (C. albicans), que se negativizó posteriormente. Evolutivamente se observó la aparición de esteatocitomas en mejillas y tronco, queratosis pilar de rodillas (fig. 2) y callosidades plantares no dolorosas, sin queratodermia palmar. Dada la sospecha clínica, se solicitó la secuenciación del gen KRT17 identificando la mutación puntual tipo cambio de sentido c.284T>C; p.Leu95Pro en heterocigosis.
Estudio molecular negativo en los progenitores, sugiriendo su aparición de novo. Actualmente el paciente tiene 4 años y mantiene tratamiento emoliente y fresado ungueal.
El principal signo de la PC es la onicodistrofia, con afectación principalmente de los dos tercios distales de la placa ungueal. Pueden asociar fragilidad, perionixis crónicas y predisposición a la sobreinfección por C. albicans o infecciones bacterianas. La positividad a Candida no excluía, por tanto, el diagnóstico de PC en nuestro paciente. Los dientes natales suelen ser la primera manifestación clínica. La hiperqueratosis palmo-plantar es la manifestación más frecuente, y suele asociar hiperhidrosis y vesículas. Otros signos son la queratosis folicular en superficies de extensión de brazos, piernas y nalgas; y leucoqueratosis oral. Cuando esta última afecta a la laringe produce ronquera y; en los casos de afectación de la membrana timpánica, sordera. Los esteatocitomas múltiples localizados en tronco, extremidades y axilas aparecen en la adolescencia2. No existe tratamiento curativo. La queratodermia palmoplantar dolorosa puede ser el principal problema. El tratamiento con queratolíticos o retinoides tópicos pueden ofrecer mejoría transitoria, indicándose retinoides orales en los casos más graves. La afectación ungueal precisa tratamiento emoliente, fresado y en ocasiones, extirpación. Los esteatocitomas múltiples y quistes pilosebáceos pueden tratarse con incisión y expresión de su contenido2.
La mutación identificada c.284T>C; p.Leu95Pro en heterocigosis en el gen KRT17ha sido previamente descrita en otro paciente afecto de PC4 y en otro registrado en The International Pachyonychia Congenita Research Registry (IPCRR)3. Un caso esporádico que no presentó dientes natales; y el segundo, un caso familiar con igual fenotipo que nuestro paciente. Existe cierta correlación fenotipo-genotipo en los 2 subtipos. El PC-1 presenta con más frecuencia leucoqueratosis oral (88% en mutaciones de KRT6A, 41% en KRT16), mientras los dientes natales y esteatocitomas son más característicos de PC-2 (76 y 67%, respectivamente en KRT17)1. Ello ha de tenerse en cuenta en la evaluación periódica de los afectados. Los dientes natales aislados tienen una prevalencia de 1/2.000-3.000 nacimientos5 y así fueron interpretados en los tíos paternos del paciente.
La caracterización molecular permite el asesoramiento genético correcto. Nuestro paciente tendrá un riesgo del 50% de transmitir la PC a su descendencia y la identificación de la mutación causal posibilita el diagnóstico prenatal o preimplantacional en el futuro. El riesgo de recurrencia en futuros hermanos, al ser un caso de novo, se estima en un máximo del 1% por la posibilidad de mosaicismo germinal en uno de los progenitores6.
A pesar de la presencia de los signos fenotípicos clave en el primer año de vida, solo el 25% de los niños son diagnosticados en este periodo, lo que conduce a un manejo y asesoramiento inadecuado de la mayoría de casos7.
A la Asociación Internacional de Paquioniquia Congénita (IPCRR-PC Project) de Salt Lake City, Utah, EE.UU. por la inclusión del paciente para su investigación molecular por parte de F.J.D. Smith y W.H.I. McLean de la Universidad de Dundee, Reino Unido.
A la familia del paciente por su colaboración y consentimiento para publicar sus datos clínicos y fotografías.
