






Los defectos congénitos de la glucosilación (CDG, por sus siglas en inglés) son enfermedades genéticas, en general multisistémicas, de herencia autosómica recesiva. Son causadas por defectos que afectan al ensamblaje, la transferencia o el procesamiento de los oligosacáridos de las proteínas u otros glucoconjugados. El CDG tipo Ib está causado por la deficiencia de la enzima citosólica fosfomanosa isomerasa (PMI), codificada por el gen MPI, que cataliza la interconversión de fructosa-6-P y manosa-6-P. Los síntomas son, fundamentalmente, gastrointestinales y hepáticos, y a diferencia de la mayoría de los pacientes con otros tipos de defectos congénitos de la glucosilación, no existe afectación neurológica. El tratamiento con manosa es muy eficaz.
Describimos el primer caso de un paciente con CDG-Ib diagnosticado en España. La enfermedad se inició clínicamente a los 6 meses con hipoglucemia, fallo de medro e hipertransaminasemia; posteriormente el paciente desarrolló una enteropatía con atrofia vellositaria subtotal detectada en la biopsia.
El paciente presentaba un porcentaje de transferrina deficiente en carbohidratos en el suero del 42 %, un patron tipo 1 en el isoelectroenfoque de la transferrina sérica, una actividad PMI en fibroblastos del 16 % y las mutaciones R219Q y R56fs en el gen MPI. El tratamiento con manosa a dosis de 1 g/kg/día en 5 dosis resultó muy eficaz, y se normalizaron tanto los parámetros clínicos como los bioquímicos.
El defecto congénito de la glucosilación Ib debería incluirse en el diagnóstico diferencial de hipoglucemias, hepatopatías, enteropatías y situaciones de hipercoagulabilidad, en ausencia de otras etiologías más comunes y, sobre todo, si se asocian varios de estos síntomas.
Congenital disorders of glycosylation (CDG) are recessively inherited multisystemic disorders resulting from several genetic defects affecting the assembly, transfer or processing of oligosaccharides onto proteins and other glycoconjugates. CDG type Ib is due to a deficiency of phosphomannose isomerase (PMI) encoded by the MPI gene. PMI catalyzes the interconversion of fructose-6-P and mannose-6-P. The clinical phenotype is characterized by gastro-intestinal and hepatic symptoms. In contrast to most CDG patients, there is no neurological affectation. It’s a mannose treatable disorder.
We report the first recognised case of CDG Ib in Spain. He presented at 6 months with hypoglycaemia, failure to thrive and hypertransaminasaemia. He subsequently developed an enteropathy with subtotal villous atrophy on biopsy.
The %CDT was very high and he presented with a type 1 pattern in transferrin isoelectric focusing. PMI activity in fibroblasts was very deficient. Mutations in MPI gene at R219Q and R56fs were found. Clinical and biochemical parameters normalised after treatment with mannose 1 g/kg/day in 5 doses.
CDG Ib should be considered in patients with hypoglycaemia, liver disease, enteropathy and hypercoagulability, in the absence of other common causes, and particularly if some of them are combined.
Los defectos congénitos de la glucosilación (CDG, por sus siglas en inglés) son un grupo de enfermedades metabólicas hereditarias multisistémicas, descritas por primera vez por Jaeken et al en 19801. Alrededor de la mitad de las proteínas del organismo están glucosiladas y éstas se clasifican, según el tipo de unión del oligosacárido (glucano) al péptido, en N-glucoproteínas, cuando el glucano se une al nitrógeno del grupo amino del aminoácido asparragina, O-glucoproteínas, si la unión es a través del oxígeno del grupo hidroxilo de los aminoácidos treonina o serina y C-glucoproteínas, si la unión es a través del carbono del grupo carboxilo del triptófano. Los defectos en la N-glucosilación pueden deberse a un trastorno en el ensamblaje, la transferencia o en el procesamiento del oligosacárido de la proteína2-4. La anomalía bioquímica común a todos ellos es la hipoglucosilación de las proteínas séricas y el método de identificación es el análisis de la transferrina por isoelectroenfoque (IEF). En la actualidad se han descrito 22 defectos genéticos diferentes, cinco de ellos afectan también a la O-glucosilación5,6, que se clasifican inicialmente en tipos I y II, según el patrón del IEF de la transferrina. El patrón tipo 1 se caracteriza por la elevación de las bandas correspondientes a las isoformas disialo y asialo-transferrina, junto con una disminución más o menos importante de la isoforma tetrasialilada de la transferrina, lo que indica una pérdida de glucanos completa, típica de los pacientes DCG-I, es decir, de los que presentan defectos en el ensamblaje del oligosacárido unido al dolicol-fosfato. El patrón tipo 2 es aquel en el que, además, pueden encontrarse aumentadas las bandas correspondientes a las isoformas trisialo y monosialo-transferrina, lo cual indica la existencia de glucanos estructuralmente anómalos, patrón que suele aparecer en los pacientes con CDG-II con defectos en el procesamiento del glucano unido a la proteína7.
En la mayoría de los casos el cuadro clínico es multisistémico, y predominan las manifestaciones neurológicas (retraso psicomotor, hipoplasia cerebelosa, retinitis pigmentaria, polineuropatía) y la coagulopatía (trombosis, hemorragias, accidentes cerebrovasculares). También son frecuentes las alteraciones gastroenterohepáticas, hormonales y cardíacas y, en ocasiones, se asocia un cuadro dismórfico8.
El CDG tipo Ib (CDG-Ib; MIM 602579) fue descrito por primera vez en 19989-11 y está causado por el déficit del enzima fosfomanosa isomerasa (PMI; EC 5.3.1.8), codificado por el gen MPI (MIM 154550)12. Esta enzima cataliza la interconversión de fructosa-6-fosfato y manosa-6-fosfato (fig. 1) y desempeña un papel importante en el mantenimiento de la reserva citoplasmática de GDP-manosa, donador de manosa en las reacciones de glucosilación.
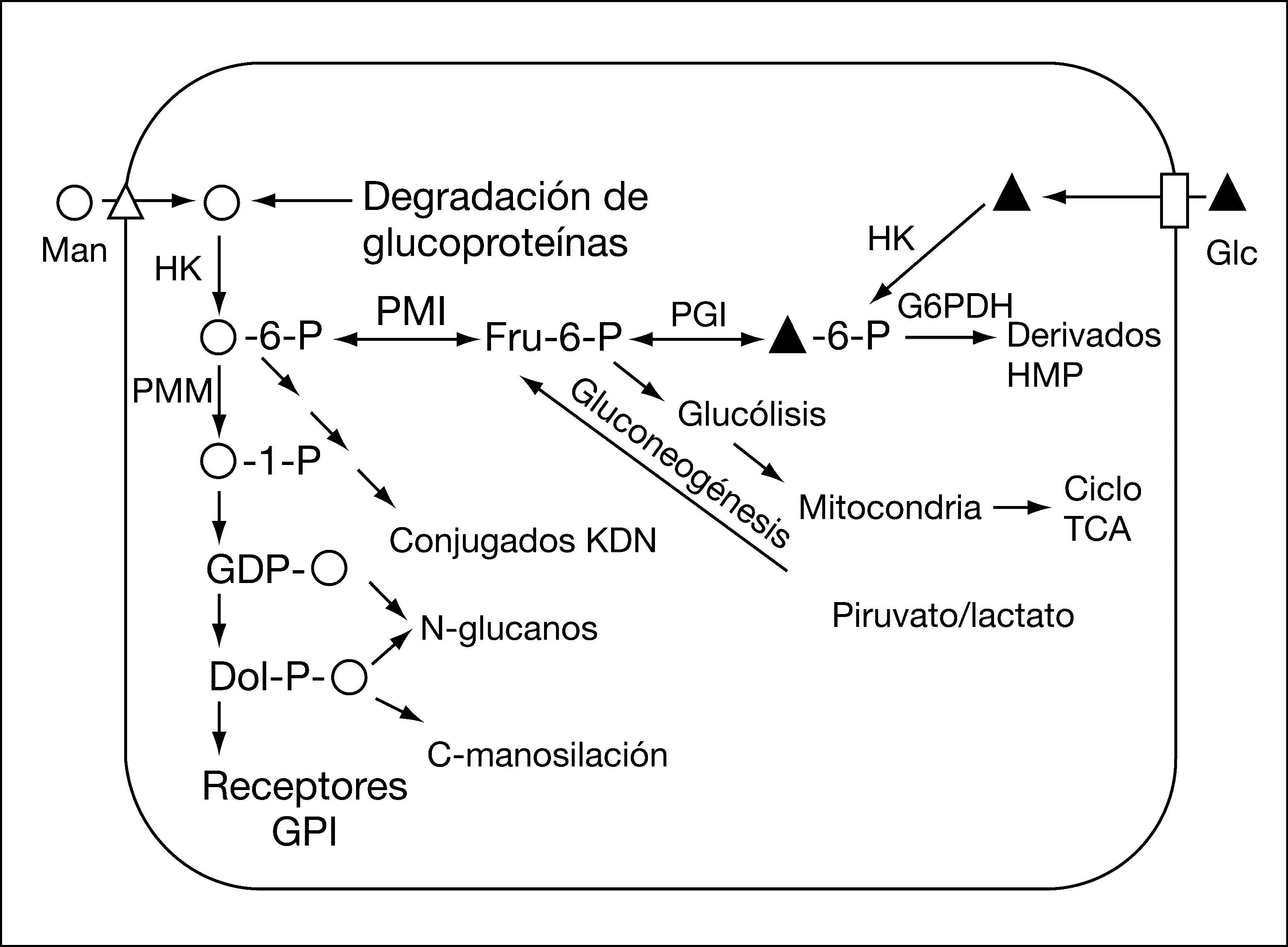
Los pacientes con CDG-Ib se diferencian del resto de los afectados de CDG en que la presentación clásica consiste en enfermedad hepática, enteropatía, trombosis venosa e hipoglucemia, sin que se hayan descrito retraso psicomotor u otras manifestaciones neurológicas13,14. A diferencia también de los otros tipos de CDG, responden muy bien al tratamiento con manosa, que se fosforilaría a manosa-6-P a través de una hexocinasa (fig. 1), obviando la vía enzimática de la PMI15. Hasta la fecha se han descrito 17 pacientes bien documentados (tabla 1).
Hallazgos clínicos de pacientes con defecto congénito de la glucosilación Ib y mutaciones identificadas en el gen MPI
| Caso | Inicio | Manifestaciones clínicas | Nucleótido | Aminoácido | Referencia bibliográfica |
| 1 | 11 meses | Enteropatía, trombosis | c.166-167insCc.656G → A | R56fsR219Q | 1025 |
| 2a | 10 meses | Vómitos recurrentes, diarrea y hepatopatía | c.457G →Ac.457G → A | R152QR152Q | 914 |
| 3a | 2 meses | Vómitos, diarrea, fibrosis hepática | c.457G →Ac.457G → A | R152QR152Q | 914 |
| 4a | 10 meses | Vómitos recurrentes, diarrea, fibrosis hepática | c.457G → Ac.457G → A | R152QR152Q | 914 |
| 5 | 3 meses | Diarrea, hipoglucemia, fibrosis hepática, coagulopatía. Fallece a los 4 años | c.304 C → Tc.413T → C | S102LM138T | 11,1320 |
| 6 | 1 mes | Fibrosis hepática, retraso psicomotor leve | c.152T → Cc.152T → C | M51TM51T | 2612 |
| 7 | 1 año | Hipoglucemia, fibrosis hepática, diarrea | c.457G → A? | R152Q? | 2612 |
| 8 | 6 meses | Fallo de medro, fibrosis hepática, enteropatía, hipoglucemia | c.391G →c.391G → A | D131ND131N | 2724 |
| 9 | 5 meses | Hipoglucemia, trombosis, enteropatía, fibrosis hepática | IVS4-1G → Cc.1252G → A | R418H | 18 |
| 10 | 2 meses | Hipoglucemia, enteropatía, coagulopatía, hepatopatía | c.764 A → Gc.1193T → C | Y255CI398T | 1320 |
| 11 | 7 meses | Enteropatía, fibrosis hepática, acidosis tubular | c.656G → Ac.748G → A | R219QG250S | 1228 |
| 12b | 2,5 años | Enteropatía, fibrosis hepática, trombosis. Fallece a los 5 años | c.636G → Ac.419T → C | R219QI140T | 22 |
| 13b | 2,5 años | Enteropatía, trombosis | c.636G → Ac.419T → C | R219QI140T | 22 |
| 14 | – | Enteropatía, hipoglucemia, coagulopatía | c.884G → Ac.884G → A | R295HR295H | 29 |
| 15 | 15 meses | Hipoglucemia, hepatopatía, coagulopatía | c.386a → Gc.457G → A | Y129CR152Q | 30 |
| 16 | – | c.466G → Ac.delG281 | E156K | 31 | |
| 17 | 4 meses | Hipoglucemia, hepatopatía, enteropatía, coagulopatía | c.656G → A166-167insC | R219QR56fs | 32Nuestro caso |
Presentamos el primer caso de CDG-Ib diagnosticado en España a la edad de 6 meses y nuestra experiencia en el tratamiento con manosa.
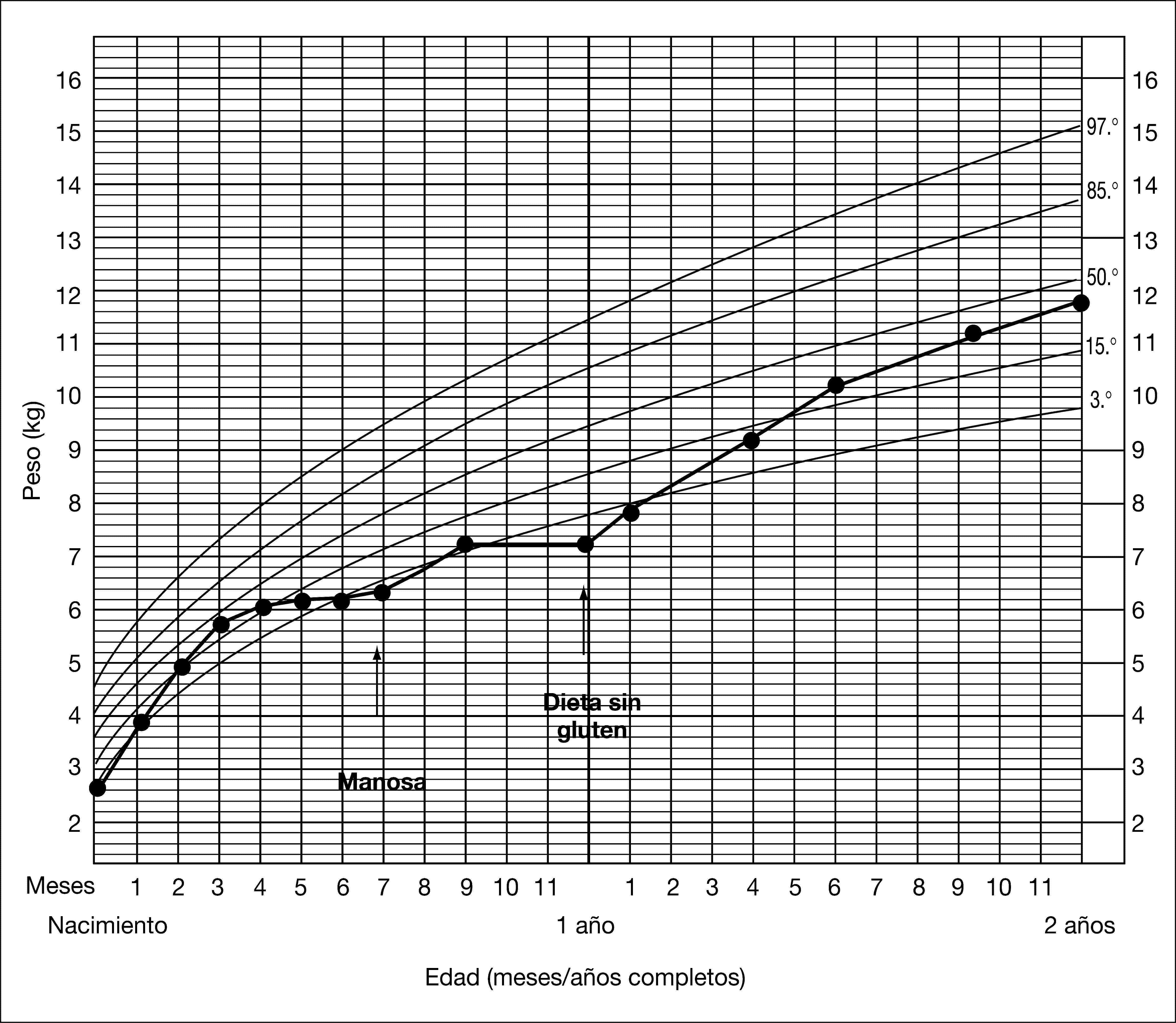
PACIENTES Y MÉTODOSCaso clínicoVarón, primer hijo de padres no consanguíneos de origen búlgaro. La madre presentó infertilidad en relación con hiperprolactinemia, por lo que recibió tratamiento con bromocriptina hasta el embarazo. La gestación, el parto y el período neonatal transcurrieron sin incidencias remarcables. El peso del recién nacido fue de 2.600g, su talla de 47,5cm y el perímetro craneal de 34cm. El cuadro clínico se inicia a los 4 meses, con estancamiento en la curva ponderal (fig. 2). Había recibido lactancia materna hasta ese momento, y a los 3 meses y medio se había introducido la fruta, presentando una buena tolerancia. A los 6 meses presentó una hipoglucemia sintomática (28mg/dl) en el curso de un cuadro infeccioso. El examen físico reveló un peso de 5.850g, (< P3), talla de 66cm (P50), signos de desnutrición, hoyuelos en la piel de ambos hombros y hepatomegalia (5cm del reborde costal). El desarrollo psicomotor era normal. En la analítica general destacaban GPT de 251 U/l y anemia microcítica. El estudio del metabolismo intermediario (aminoácidos, ácidos orgánicos, acilcarnitinas, lactato, piruvato y amonio) fue normal, objetivándose un porcentaje de transferrina deficiente en hidrato de carbono (%CDT) en suero muy alto (42 %) y un patrón de isoformas de transferrina patológico tipo 1 (fig. 3, carril 2). Estos hallazgos sugerían la existencia de un CDG. En el estudio de coagulación se observó un descenso de proteínas S y C, antitrombina III y factor XI. Las sustancias reductoras en orina fueron negativas y el estudio del gen Aldolasa B fue normal. Debido a que el paciente no tenía síntomas neurológicos, se sospechó un CDG tipo Ib que posteriormente se confirmó por estudios enzimáticos y genéticos. A los 7 meses de vida se inició el tratamiento con manosa a 150mg/kg en 4 dosis, con lo que mejoró la curva de peso en los meses siguientes (fig. 2) y descendió el %CDT al 24 %, aunque persistía el patrón tipo 1 de la transferrina (fig. 3, carril 3). A los 9 meses se introduce el gluten en la dieta. A los 12 meses inició una diarrea prolongada con pérdida de peso, hipoproteinemia, hipoalbuminemia y anticuerpos antigliadina IgA positivos con anticuerpos antitransglutaminasa normales. En este momento se retiró la manosa dado que incrementaba el número de deposiciones. Se realizó biopsia duodenal en la que se observó una atrofia subtotal de vellosidades intestinales. Se inició dieta sin gluten y se reintrodujo la manosa a dosis bajas, con una evolución satisfactoria respecto a la curva de peso (fig. 2) y diarrea, pero persistiendo un %CDT muy alto (30%), con patrón muy patológico de la transferrina (fig. 3, carril 4), y descenso de antitrombina III (AT III) e hipertransaminasemia (GPT 293 U/l). Se incrementó la manosa a dosis de 200mg/kg en 5 dosis (1g/kg/día) y 1 mes después la GPT descendió a 123 U/l. A los 6 meses, la GPT era de 32 U/l, el estudio de coagulación era normal y el %CDT era del 6%, con una evidente mejora del perfil de glucosilación de la transferrina (fig. 3, carril 5). El estudio de los antígenos de histocompatibilidad HLA no demostró la existencia de factores de riesgo de enfermedad celíaca. Se efectuó una segunda biopsia tras 20 meses de dieta sin gluten y en tratamiento con manosa que fue normal, así como el linfograma de la mucosa intestinal, con una proporción de linfocitos gamma/delta no compatible con enfermedad celíaca, por lo que se volvió a introducir el gluten. A los 3 años y medio, el paciente está asintomático, sigue una dieta libre y tiene un desarrollo físico y psicomotor normales.
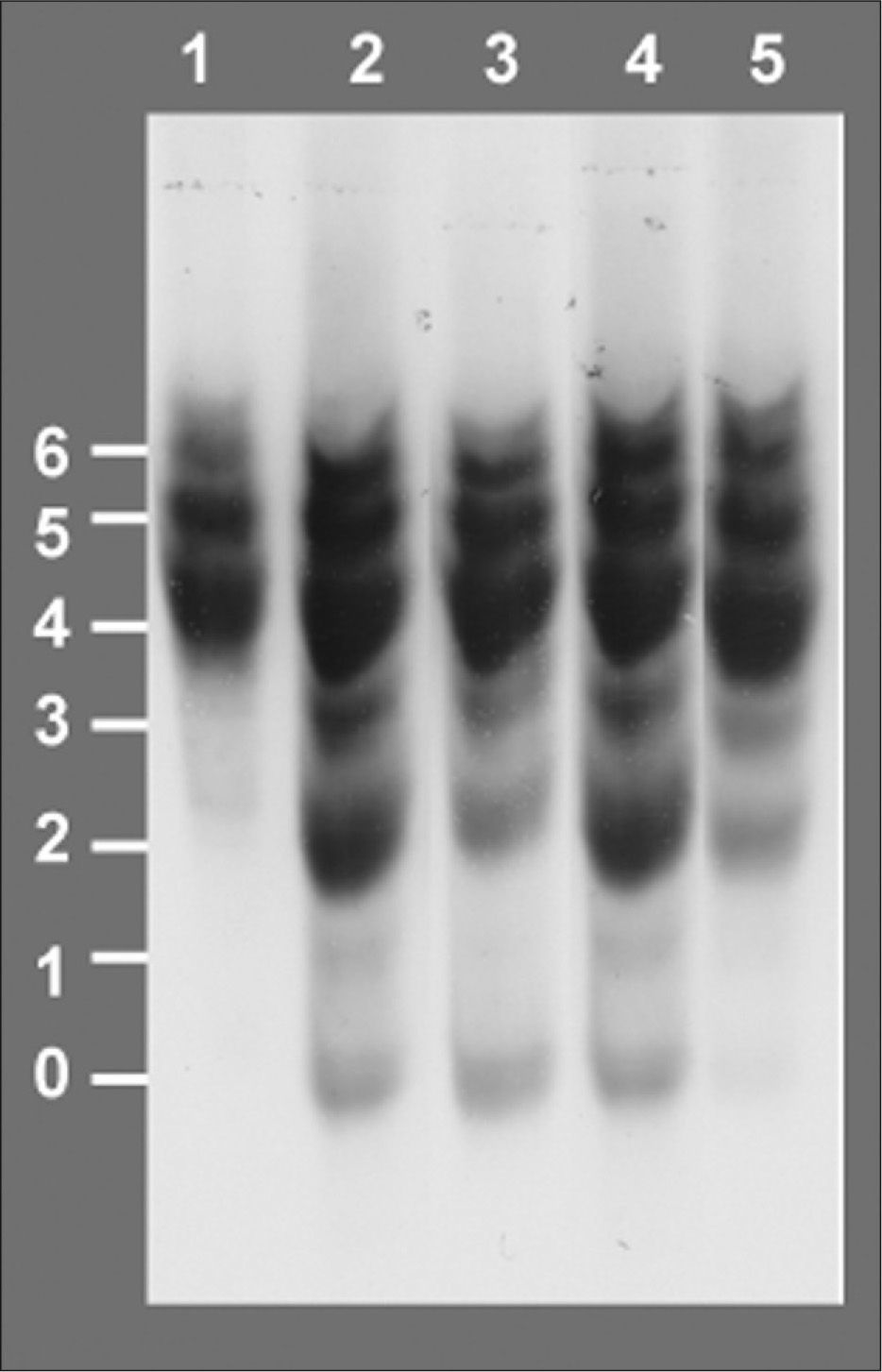
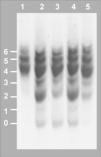
Isoelectroenfoque de la transferrina sérica. Los números a la izquierda indican el grado de glucosilación de las diferentes isoformas de la transferrina, siendo las bandas 0, 1 y 2 las que corresponden a las formas hipoglucosiladas a-, mono y di-sialotransferrina, respectivamente. Carril 1: control. Carril 2: paciente al diagnóstico, episodio de hipoglucemia, desnutrición, hepatomegalia e hipertransaminasemia, 42% de %CDT. Carril 3:paciente en tratamiento con manosa a dosis de 150mg/kg en 4 dosis, mejoría en la curva de peso, 24 % de %CDT. Carril 4, paciente tras presentar diarrea prolongada, hipoalbuminemia, pérdida de peso y anticuerpos antitransgliadina positivos, dieta sin gluten, administración irregular de manosa, 30 % de %CDT. Carril 5: paciente 6 meses después de incrementar la dosis de manosa a 1g/kg/día en 5 dosis, curva de peso ascendente, transaminasas y estudio de coagulación normal, 6 % de %CDT.
El %CDT se determinó en suero mediante un inmunoanálsis turbidimétrico, utilizando el equipo %CDT TIA BIO-RAD Microtiter plate version. Este ensayo mide la suma de las isoformas con 0, 1 o 2 residuos de ácido siálico de la transferrina en proporción a la cantidad de transferrina total sérica circulante16.
Isoelectroenfoque (IEF) de la transferrinaLa muestra de suero saturada de hierro se separa en un gel de agarosa que contiene anfolinas (pH 4,6-8) en una unidad de electroforesis Multiphor II (Amersham Bioscience). Después de la transferencia a un filtro de nitrocelulosa, las isoformas de la transferrina correspondientes a los diferentes contenidos de ácido siálico se detectaron con los anticuerpos específicos antitransferrina16.
Determinación de las actividades enzimáticas fosfomanosa isomerasa (PMI) y fosfomanosa mutasa (PMM)Las actividades enzimáticas de las enzimas citosólicas PMI y PMM se determinaron a 30°C en fibroblastos de piel cultivados según una modificación del método de Van Schaftingen y Jaeken17.
Análisis de mutaciones del gen MPIPara la extracción del ARN total a partir de fibroblastos de piel del paciente se utilizaron TriPure de Ambion y su posterior conversión en ADNc (1μg de ARN total) se hizo con el equipo SuperScriptIII de Invitrogen. La extracción del ADN genómico a partir de fibroblastos se realizó mediante fenolizaciones, y de la sangre impregnada en papel de los padres mediante el equipo Generation DNA Purification Systems de Gentra Systems. Se diseñaron los primers específicos para la amplificación del ADNc y de los 7 exones del gen MPI basándose en la secuencia descrita en EMSEMBL. La purificación de los productos obtenidos se realizó mediante los sistemas comerciales SpinClean PCR Purification Kit de Messenger of Biotechnology o QIAEX II Gel extration Kit de QIAGEN. Para la separación alélica e identificación de la inserción se clonó el exón 3 del gen MPI en el vector de clonaje TOPO (Invitrogen). La secuenciación cíclica directa se llevó a cabo utilizando el equipo BigDey Terminator v3.1 Cycle Sequencing de Applied Biosystems.
RESULTADOSA los 6 meses de vida se realizó un estudio metabólico con el fin de descartar enfermedades hereditarias causantes de hipoglucemia, evidenciándose un valor del %CDT en suero muy elevado (42%, valor control < 3%). El patrón de isoformas de la transferrina de dicho suero por isoelectroenfoque fue también muy patológico (tipo 1), y se detectó un gran aumento de las bandas correspondientes a las isoformas di-y a-sialiladas de la transferrina (fig. 3, carril 2). Estos resultados eran compatibles con un CDG tipo I, es decir, con un defecto de glucosilación que afecta al ensamblaje del oligosacárido estándar y su transferencia a la proteína. Se excluyeron causas secundarias de hipoglucosilación como galactosemia o intolerancia a la fructosa. Se determinaron en fibroblastos de piel las actividades PMM, cuya deficiencia es la causa más frecuente de un CDG y PMI, detectándose una actividad PMI deficiente (2,1mU/mg proteína; valor control = 12,8 ± 2,7), lo que confirmaba el diagnóstico de un CDG tipo Ib. El análisis mutacional reveló la presencia de dos mutaciones en trans en el ADNc del gen MPI (fig. 4). Ambas mutaciones se confirmaron en ADN genómico del paciente y de sus padres. En el alelo heredado de su madre se identificó la mutación c.656G > A, que da lugar al cambio del aminoácido arginina por glutamina en la posición 219 de la proteína (R219Q). La mutación heredada del padre (R56fs) consiste en una inserción de una citosina entre la posición 166 y 167 del exón 3 (166-167insC) que da lugar a un codón de parada en la posición 62 de la proteína.
DISCUSIÓN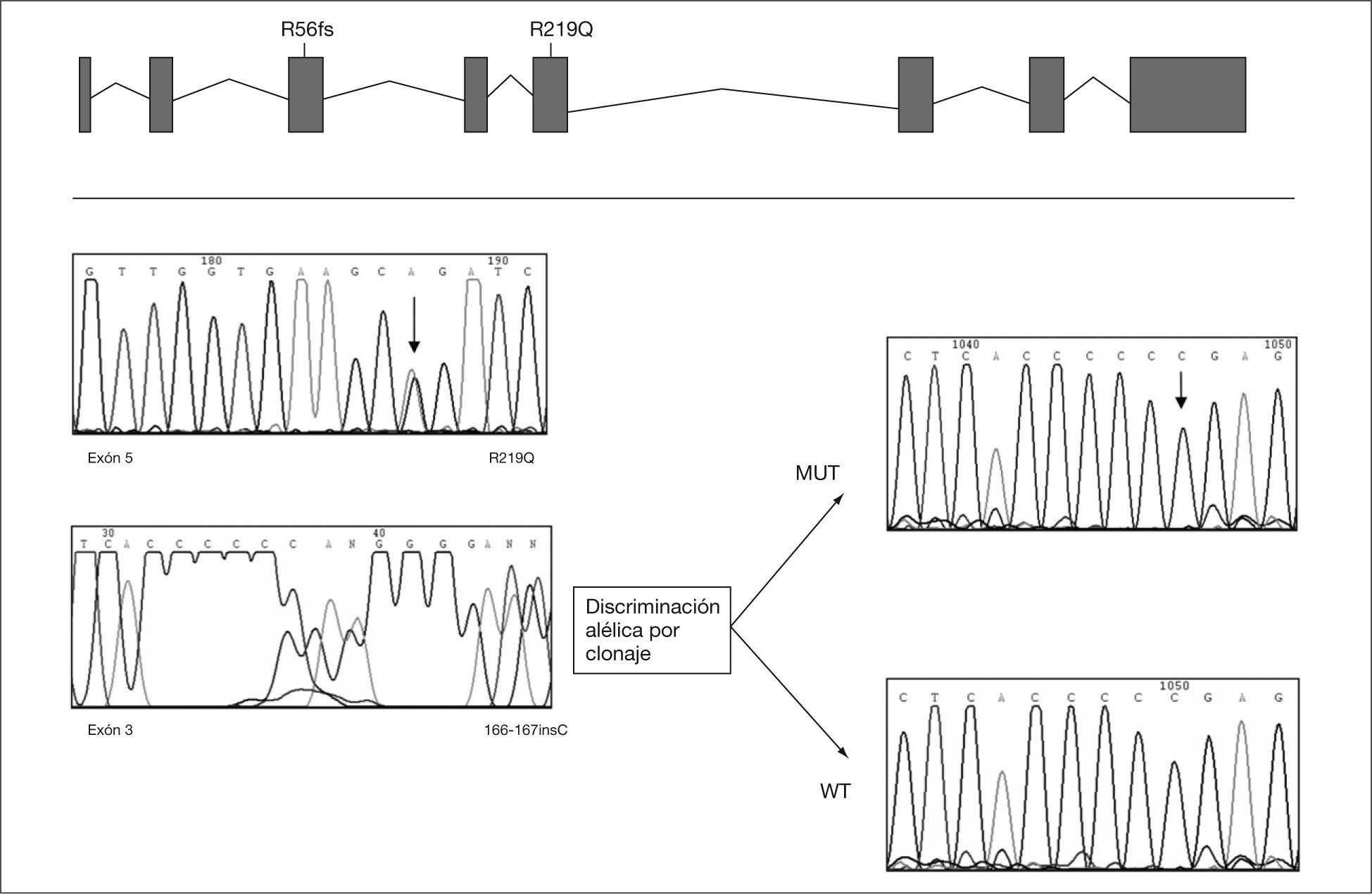
La deficiencia de la enzima citosólica fosfomanosa isomerasa, que cataliza el primer paso en la vía de síntesis de GDP-manosa (fig. 1), causa el CDG tipo Ib. La presentación clínica es eminentemente hepática y gastroenterointestinal sin asociarse con retraso psicomotor ni con otras manifestaciones neurológicas, como ocurre en la mayoría de los CDG. Probablemente esta diferencia en las manifestaciones clínicas esté relacionada con que, al estar indemne la vía de la hexocinasa, se sintetiza alguna cantidad de GDP-manosa, donador de manosa en el citosol; por otra parte, se ha descrito que probablemente durante el embarazo la manosa materna sea suficiente para mantener las reacciones de glucosilación en el feto que permiten el desarrollo normal del sistema nervioso18.
La madre del paciente presentó infertilidad en relación con hiperprolactinemia, hallazgo que no hemos encontrado referido en pacientes con CDG-Ib, pero sí en pacientes adultas con CDG tipo Ia19. También se han descrito fluctuaciones en las concentraciones de prolactina en la primera niña cuyo caso se describió1.
El diagnóstico del paciente fue muy precoz debido a que la forma de presentación, con hipoglucemia sintomática grave, hepatopatía y retraso ponderal en un lactante de 6 meses, condujo a efectuar un estudio metabólico para descartar defectos en la betaoxidación de ácidos grasos, neoglucogénesis y glucogenosis. También se excluyó la intolerancia hereditaria a la fructosa. No se pudo hacer un estudio hormonal para comprobar si la hipoglucemia estaba en relación con hiperinsulinismo como en otros casos descritos13,18,20. El gran aumento del %CDT en suero, de hasta 14 veces el valor normal y el patrón patológico de las isoformas de la transferrina orientó el diagnóstico hacia un CDG, y aunque estas alteraciones bioquímicas no permiten distinguir entre los diferentes subtipos de CDG tipo I, la ausencia de signos malformativos, como acumulación anómala de grasa o mamilas invertidas, o de signos neurológicos, sugería que el paciente podía tener una forma tipo Ib. El diagnóstico se confirmó demostrándose un déficit de la actividad PMI en fibroblastos y dos mutaciones en trans ya descritas en el gen MPI10. El cambio R219Q es un cambio conservado en diferentes organismos, C. albicans, C. elegans, ratón y humano, y ha sido expresado en células COS-7, mostrando una actividad PMI prácticamente indetectable10, lo que confirma su patogenicidad. La otra mutación R56fs da lugar a un codón de parada en la posición 62 de la proteína, que presumiblemente sería inactiva.
Además de hipoglucemia, el paciente presentó desde el principio una hepatopatía que se asumió que estaba relacionada con su enfermedad de base. No se pudo efectuar biopsia hepática dados los trastornos de coagulación que presentaba. El hallazgo histológico más frecuente encontrado en estos pacientes es la fibrosis periportal9,11,18,21,22, aunque también se han encontrado esteatosis11,20, cirrosis20, proliferación de canalículos biliares11 y hallazgos similares a los complejos de Meyenburg21. En el estudio ultraestructural se observan inclusiones lisosomales21.
Las manifestaciones digestivas de los pacientes con CDG son muy frecuentes20,21,23, e incluyen fallo de medro y diarrea con hipoproteinemia e hipoalbuminemia, con esteatorrea o sin ella. Se han descrito también una gran variedad de hallazgos patológicos en la biopsia intestinal, como inflamación linfoplasmocitaria del corion, linfangiectasias, acumulaciones intravacuolares de lípidos y atrofia vellositaria parcial. Boyer et al20 señalan que estos hallazgos sugieren la existencia de varios mecanismos etiopatogénicos como inflamación, alteraciones en el transporte de lípidos o aumento de permeabilidad intestinal20. Nuestro paciente presentó al año de vida una diarrea prolongada con hipoproteinemia y anticuerpos antigliadina positivos. Se observó una atrofia vellositaria subtotal y ante la duda de si se trataba de una enfermedad celíaca o de si los anticuerpos antigliadina eran la consecuencia de un aumento de la permeabilidad intestinal, se inició una dieta sin gluten con evolución satisfactoria. Posteriormente, el estudio de HLA, el linfograma de la mucosa duodenal y la ausencia de síntomas tras la reintroducción del gluten descartaron una enfermedad celíaca.
Con respecto al tratamiento con manosa, la dosis inicial de 150mg/kg cuatro veces al día no fue lo suficientemente eficaz, ya que si bien desapareció la sintomatología, persistieron las anomalías bioquímicas y algunos meses después se manifestó la enteropatía. No obstante, no se puede afirmar que se le estuviera administrando correctamente, ya que no se monitorizaron los valores de manosa. Tras incrementar la dosis a 200mg/kg cinco veces al día se normalizaron todos los parámetros bioquímicos a los 6 meses de tratamiento. La dosis y el esquema de administración recomendados en la actualidad son de 150–200mg/kg cinco veces al día24,25.
El pronóstico a largo plazo con tratamiento o sin él no se conoce bien todavía, ya que es una entidad recientemente caracterizada. El único caso adulto descrito es el de una mujer de 33 años cuyo hermano había fallecido a los 5 años de vida por una hemorragia intestinal. La paciente, sin embargo, se había mantenido asintomática excepto por dos brotes de enteropatía y trombosis a los 2 y 3 años22.
Como conclusión, queremos señalar la importancia de incluir los CDG tipo Ib en el diagnóstico diferencial de los cuadros de diarrea crónica, fallo de medro, hepatopatía, hipoglucemia y situaciones de hipercoagulabilidad, en ausencia de otras causas más comunes que los justifiquen y sobre todo si se asocian varios de estos síntomas. Por último, también queremos señalar la utilidad de la medida del %CDT en suero como marcador bioquímico para el diagnóstico y seguimiento de los CDG.
FinanciaciónEl trabajo ha sido financiado por el proyecto P1040791 del Fondo de Investigaciones Sanitarias (Instituto Carlos III) y el proyecto del Sexto Programa Marco de la Unión Europea Euroglycanet: LSHM-CT2005-512131.
La ayuda institucional de la Fundación Ramón Areces al Centro de Biología Molecular Severo Ochoa.

Anales de Pediatría sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas
- Inicio
- Todos los contenidos
- Publique su artículo
- Acerca de la revista
- Métricas