



El síndrome de deficiencia del transportador de glucosa tipo 1 (Glut 1) es un defecto genético causado por mutaciones en el gen SLC2A1 (1p35-p31.3)1. Se caracteriza por hipoglucorraquia y disminución del cociente de glucosa en el líquido cefalorraquídeo (LCR)/glucosa en sangre, y generalmente se manifiesta como una encefalopatía epiléptica de comienzo precoz refractaria al tratamiento con fármacos antiepilépticos (FAES). Con frecuencia el diagnóstico se demora, lo que puede conllevar secuelas neurológicas graves.
Presentamos el caso de una niña de 33 días de vida que ingresa por crisis convulsivas. Es la segunda hija de padres no consanguíneos. Amenaza de aborto en el primer trimestre. Parto y período neonatal sin incidencias. En las horas previas presenta episodios de hipotonía generalizada, cianosis facial y desconexión ambiental de una duración aproximada de 1min. A su llegada al hospital, la exploración física no revela anomalías. Durante el primer día se repiten las crisis en dos ocasiones y se inicia tratamiento con fenobarbital. El hemograma, bioquímica sanguínea y una ecografía cerebral no demostraron anomalías. El LCR en dos ocasiones mostró una glucorraquia de 15mg/dl, sin aumento de proteínas ni celularidad. Las cifras de lactato en el LCR se encontraban en el rango bajo de la normalidad. La glucemia antes de la punción lumbar fue de 93mg/dl, con un cociente glucorraquia/glucemia de 0,16. Una resonancia magnética cerebral mostró una pequeña lesión bilateral, en la cápsula externa hipointensa en T1 e hiperintensa en T2. En el surco central parietal se observa una pequeña zona de hiperseñal. La tomografía por emisión de positrones (PET) a nivel cerebral con fluorodesoxiglucosa (fig. 1) reveló una corteza cerebral con captación de glucosa globalmente disminuida, con un predominio aparente de la señal en los ganglios de la base e hipocaptación en los tálamos, sin lesión capsular acompañante detectable, junto con una disminución de la señal cerebelosa. Ante la sospecha de una deficiencia de Glut 1, se retiró el fenobarbital y se inició dieta cetogénica con un aporte de grasas del 60 %. El electroencefalograma (EEG) evidenció elementos agudos alternantes de predominio izquierdo. Tras la retirada del fenobarbital, no se observaron cambios significativos en la glucorraquia. En los primeros 4 meses de vida presentó episodios esporádicos de cese de actividad y fijación de la mirada de breve duración que no han recurrido tras el incremento del aporte de lípidos al 70 %. Con 14 meses, su desarrollo es normal y el crecimiento del perímetro craneal se mantiene en el percentil 50. Un nuevo análisis del LCR que reveló una glucosa de 29mg/dl. El estudio genético está en curso.
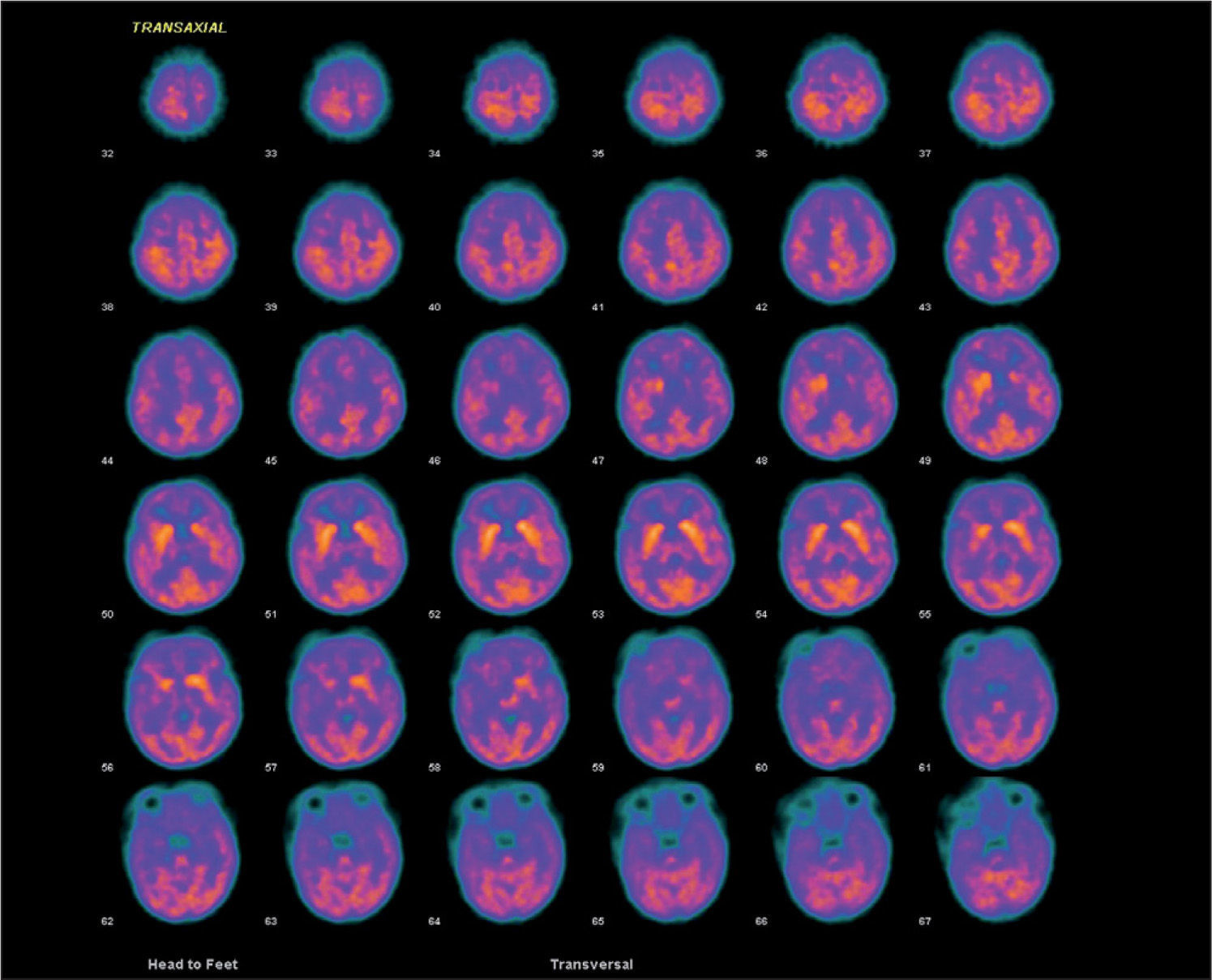
Nuestro caso se manifestó por convulsiones precoces, principal expresión clínica inicial del síndrome de deficiencia de Glut 12–4, aunque también puede cursar como un retraso mental asociado a disartria y ataxia intermitente sin epilepsia4,5 o, con menor frecuencia, por retraso en el desarrollo, coreoatetosis y distonía2,6. Se han descrito crisis generalizadas, tónicas o clónicas, mioclónicas, parciales, ausencias o atónicas como las que presentó nuestro paciente, así como crisis no clasificadas2,3,7. El EEG es inespecífico y con frecuencia no muestra anomalías7. Evolutivamente, la RM cerebral puede mostrar una hipotrofia cerebral y otras anomalías inespecíficas2,6 o de significado desconocido, como ocurrió en este caso. Una hipoglucorraquia con glucosa < 40mg/dl y un cociente de glucosa en LCR/glucosa en plasma ≤ 0,33 es altamente sugestivo de esta entidad en ausencia de infección o hemorragia subaracnoidea8. Los bajos valores de lactato en el LCR apoyan el diagnóstico. La concentración de glucosa en el LCR en el caso descrito es inferior a lo reflejado en la bibliografía2,6, y podría estar relacionado con una grave disminución del transporte de glucosa a través de la barrera hematoencefálica, que depende casi exclusivamente de Glut 1. El diagnóstico puede confirmarse mediante pruebas funcionales (captación de glucosa en eritrocitos o PET) y moleculares. La PET con aparente hipometabolismo difuso en la corteza y tálamo e hipermetabolismo aparente o relativo en los ganglios de la base es característica de esta entidad9. Es importante confirmar el diagnóstico del déficit de Glut 1, pues se han descrito formas de hipoglucorraquia transitoria en niños10. El tratamiento con FAES es ineficaz e incluso algunos fármacos pueden agravar el proceso2,3. El tratamiento con dieta cetogénica es efectivo en el control de las crisis2,3, aunque para algunos autores la utilidad en los déficits cognitivos es escasa2,3. Nuestra paciente con 14 meses de edad presenta un neurodesarrollo acorde con su edad cronológica. Consideramos que debe descartarse esta entidad ante una epilepsia de comienzo precoz y escasa respuesta a FAES, y debe incluirse en el diagnóstico diferencial del retraso mental no filiado.
