



Se presenta un caso de deficiencia de holocarboxilasa sintetasa con actividad piruvato carboxilasa normal en linfocitos en una niña de 8 años con clínica de intoxicación y sin la clásica afectación dermatológica. La identificación de 3 cambios nucleotídicos en el gen HCLS, habiendo sido descrito como mutación patogénica solo uno de ellos, podría estar relacionada con una variante leve de la enfermedad que explicaría la presentación inusual más allá de la época de lactante. El tratamiento con biotina a 40mg/día, junto con dieta controlada en proteínas, permite un crecimiento físico y un desarrollo psicomotor normales para su edad.
We report a case of holocarboxylase synthetase deficiency with normal pyruvate carboxylase activity in the lymphocytes of an 8 year-old girl with clinical toxicity without the classic dermatological involvement. The identification of three nucleotide changes in the holocarboxylase synthetase (HLCS) gene, only one of them described as a pathogenic mutation could be related to a slight variant of the disease that would explain the unusual presentation beyond the age of infant. Treatment with biotin at 40mg/day with protein controlled diet allows normal physical growth and psychomotor development for their age.
La holocarboxilasa sintetasa (HLCS: EC 6.3.4.10) es una enzima que cataliza la unión covalente de biotina a las 2 isoformas de la acetil-CoA carboxilasa citosólica (ACC1 Y ACC2) implicadas en la biosíntesis de ácidos grasos y a las 3 carboxilasas mitocondriales: piruvato carboxilasa (PC), enzima clave de la gluconeogénesis, propionil-CoA carboxilasa (PCC) y la β-metilcrotonil-CoA carboxilasa (MCC), enzimas implicadas en el catabolismo de aminoácidos (fig. 1)1. La deficiencia de HLCS (OMIM 253270) es un trastorno de herencia autosómica recesiva que conlleva un déficit múltiple de las carboxilasas indicadas. Los pacientes con deficiencia de HLCS se presentan habitualmente en el período neonatal o en la época de lactante con acidosis metabólica, hiperamoniemia, exantema, alopecia, hipotonía, convulsiones, rechazo de tomas, letargia y/o coma. El bloqueo enzimático da lugar a la aciduria/emia orgánica característica de la enfermedad. La presentación en el niño mayor es excepcional. Se presenta un caso que comenzó a los 8 años, en el que se demostró un déficit de 2 de las 3 carboxilasas mitocondriales en linfocitos.
Caso clínico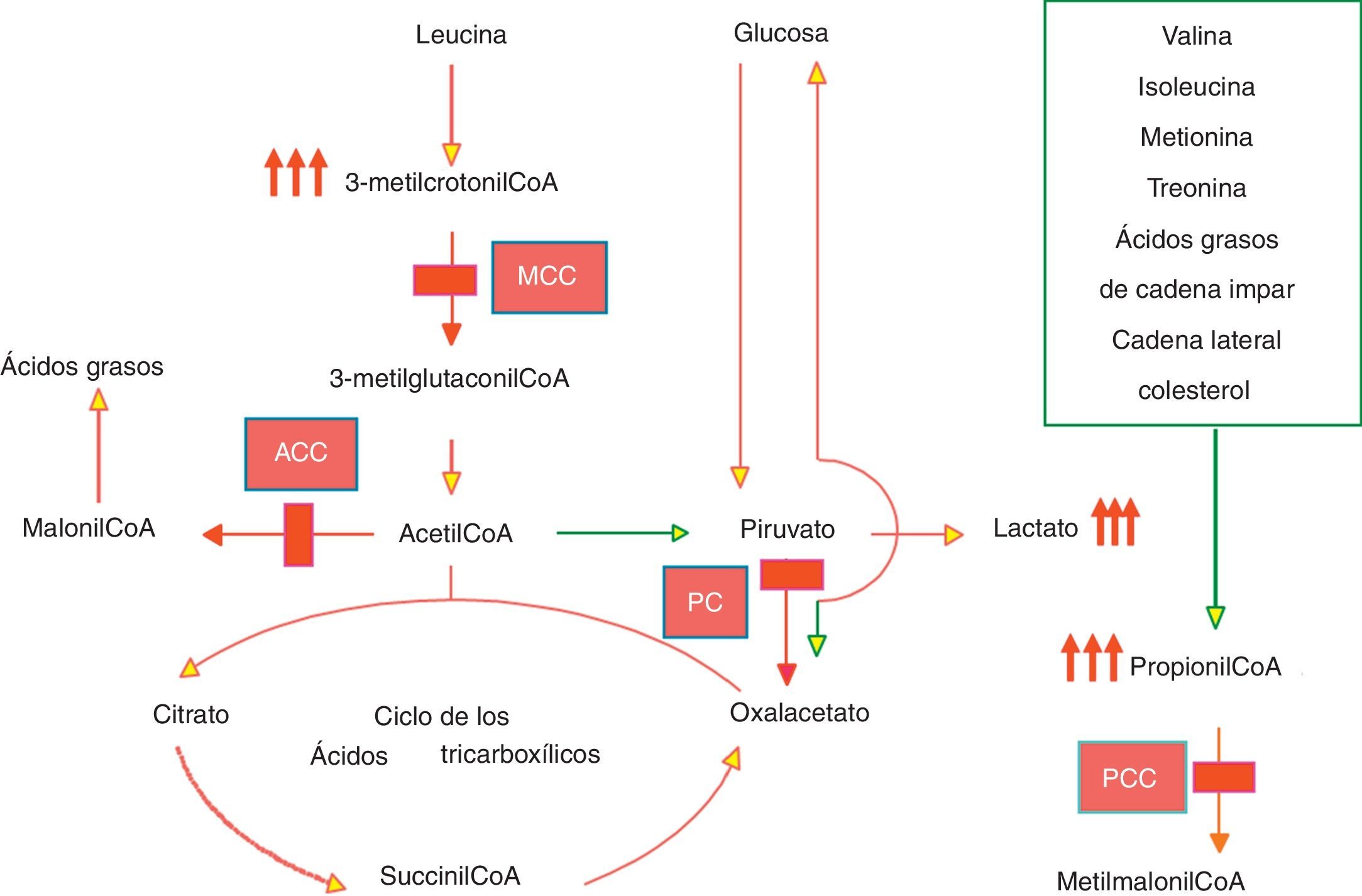
Paciente de 9 años con antecedentes de 2 episodios en el último año de ingreso en otro hospital por hipoglucemia cetósica, vómitos y acidosis metabólica (glucemia 48mg/dl, pH 7,24, bicarbonato 13,2mmol/l, EB –12,8mmol/l, lactato 2,9mmol/l (v.n. < 2,5), amonio 40 μmol/l (v.n. < 50) y acetoacetato orina > 160mg/dl. Aminoácidos séricos, normales. Ácidos orgánicos en orina: aumento de ácido 3-hidroxiisovalérico (252mmol/mol creat.; v.n. < 50), β-metilcrotonilglicina (129mmol/mol creat.; v.n. < 20), metilcitrato (26 mmol/mol creat.; v.n. < 12) y ácido 3-hidroxipropiónico (53 mmol/mol creat.; v.n. < 20). Los niveles en sangre de 3-OH-isovalerilcarnitina (C5OH) y propionilcarnitina (C3) también estaban elevados (7,3 μmol/l; v.n. < 0,52, y 8,3 μmol/l; v.n. < 3,7, respectivamente). La actividad biotinidasa sérica fue normal. Antecedentes personales: hija de padres no consanguíneos, nacida a término, adecuada a la edad gestacional, con crecimiento físico y desarrollo psicomotor normales. Peso 29,7kg (percentil 10-25) y talla 134cm (percentil 10-25). La exploración cutánea es normal. El pelo es algo ralo y quebradizo. Se le prescribe dieta con 1g/kg día de proteínas y suplementos de carnitina, y se remite a la Unidad de Metabolopatías.
Tras dicho tratamiento persiste la excreción ligeramente aumentada de metilcitrato en orina y C3 en plasma. Acidemia láctica normal. Aminoácidos en plasma y orina, normales. Se determinan las actividades carboxilasas mitocondriales en linfocitos, demostrándose PC normal (136pmol/min/mg prot; v.n. 70±32), mientras que PCC y MCC son muy deficientes (80pmol/min/mg prot; v.n. 1.099±373 y 76pmol/min/mg prot; v.n. 404±142, respectivamente). Tras administrar 20mg/día de biotina, se normalizan las actividades PCC y MCC, así como la excreción de ácidos orgánicos en orina y de C3 en plasma.
En el análisis del gen HLCS en ADN de la paciente se identifican 3 cambios nucleotídicos. El estudio en ADN de los padres muestra que el alelo paterno porta el cambio c.1648G>A (p.Val550Met), mutación previamente descrita en el Human Gene Mutation Database. El alelo materno porta los cambios c.1921G>A (p.Val641Met), descrito como polimorfismo en la base de datos 1000 Genomes y c.1696G>A (p.Val566Met), no descrito previamente, ambos de efecto funcional desconocido.
Al año de seguimiento, presenta un episodio de náuseas y somnolencia, sin acidosis metabólica ni hipoglucemia. En el análisis de ácidos orgánicos en orina de dicho episodio destacan un ligero aumento de ácido láctico (1.219mmol/mol creat.; v.n. 5-113) y cetonuria (ácidos 3-OH-butírico 2.339 mmol/mol creat; v.n. 2-17, y acetoacético 750 mmol/mol creat; v.n. 0-7). El análisis de acilcarnitinas en sangre es normal. Se decide aumento de aporte de biotina a 40mg diarios, continuando con alimentación controlada en proteínas (1,14g/kg de proteínas naturales y 0,25g/kg día de proteínas sintéticas limitadas en leucina (X-Leu Maxamaid®)), con lo que continúa asintomática durante los 3 años de seguimiento. El crecimiento físico, el desarrollo psicomotor y la evaluación nutricional durante el seguimiento son normales.
ComentariosEl diagnóstico de sospecha de deficiencia de HLCS se realizó gracias al estudio de ácidos orgánicos en la recogida inicial de orina. En este sentido, debe enfatizarse el papel de la recogida de muestras de sangre y orina en cualquier ámbito asistencial2. En los casos de diagnóstico en los primeros días o meses de vida suelen ser las lesiones dermatológicas3–5 o las alteraciones neurológicas, en forma de retraso psicomotor o convulsiones refractarias6 los motivos principales de consulta. Sin embargo, en los casos de presentación de los 5 a los 8 años, la clínica predominante son los episodios de intoxicación con vómitos y acidosis metabólica7,8, al igual que en el caso presentado. El diagnóstico final se estableció al demostrar la normalización «in vivo» de las actividades PCC y MCC como respuesta a la administración de biotina, junto con una actividad biotinidasa en suero normal, cuyo déficit puede también resultar en una deficiencia múltiple de carboxilasas. El hallazgo de una actividad PC en linfocitos normal antes de la administración de dosis farmacológicas de biotina al paciente fue sorprendente. Los pacientes descritos en trabajos precedentes tienen la actividad PC en células (linfocitos o fibroblastos) reducida en ausencia de biotina, aun en aquellos con formas de presentación tardía9,10. Está descrito que la PCC y la MCC podrían ser más sensibles a concentraciones deficientes o limitantes de biotina que la PC y la ACC, enzimas muy reguladas, ya que son clave en procesos biológicos primordiales, como son la gluconeogénesis y la síntesis de ácidos grasos11. Por esta razón, los indicadores más sensibles de una deficiencia o limitación de biotina son los metabolitos urinarios ácido 3-hidroxiisovalérico, β-metilcrotonilglicina y metilcitrato, metabolitos que se acumulan en las deficiencias de MCC y PCC11. En el caso presentado, no conocemos la concentración de biotina sanguínea cuando se realizó la primera determinación de carboxilasas en linfocitos, y podría especularse con que fuera suficiente para mantener una actividad PC normal, lo que se correspondería con la ausencia de acidemia láctica y la forma leve de la enfermedad que presenta. Desafortunadamente, no se pudieron realizar ensayos de reactivación de las carboxilasas y su dependencia a dosis variables de biotina en fibroblastos.
La biotina es una vitamina hidrosoluble que actúa como grupo prostético de las carboxilasas. Los pacientes con deficiencia de HLCS responden al empleo de la biotina por 3 posibles mecanismos de acción: el aumento de afinidad enzimático, el aumento de la transcripción del gen HLCS, con lo que se sintetiza más HLCS, o la falta de saturación de la enzima a concentraciones fisiológicas de biotina, permitiendo un aumento de actividad cuando se aumenta la concentración12. La dosis de biotina empleada en los casos publicados ha sido desde 1,2mg hasta 100mg diarios7,13,14. Además, dado que 2 carboxilasas deficientes están implicadas en el catabolismo de los aminoácidos, parece razonable una restricción moderada proteica15.
En el caso presentado, tras la restricción proteica inicial se ha ido aumentando la dosis de biotina de 20 a 40mg diarios, a la vez que se aumentaba el aporte proteico, comprobándose en todo momento la ausencia de ácidos orgánicos y acilcarnitinas patológicas y un desarrollo psicomotor normal con rendimiento escolar adecuado a su edad.
En resumen, resaltamos la singularidad de esta paciente con una expresión clínica leve de la enfermedad asociada a actividad PC normal en células periféricas y 3 cambios en el gen HLCS, 2 de ellos con efecto funcional desconocido.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
