


Introducción
La dermatomiositis juvenil es una enfermedad poco frecuente en niños (incidencia estimada de 9 casos/106 niños/año) aunque con importantes consecuencias familiares y sociosanitarias. Tiene una presentación bimodal con un pico a los 5-6 años de edad (sin predominio de sexo) y otro a los 10 años (predominio femenino)1.
El diagnóstico se establece con los criterios de Bohan y Peter1,2. El diagnóstico definitivo requiere la presencia de tres criterios de los cuatro enumerados (debilidad muscular proximal simétrica, elevación de enzimas musculares, electromiograma típico de miositis y biopsia muscular compatible con miositis inflamatoria), independientemente del exantema y la exclusión de otras enfermedades del tejido conjuntivo.
Se presenta el caso de un paciente diagnosticado de lupus eritematoso discoide que presentó en su evolución contracturas musculares y calcinosis.
Observación clínica
Niño de 5 años y 5 meses de edad que había sido diagnosticado de lupus discoide a partir de la biopsia de lesiones papulares en mejillas y nudillos de las manos y tratado con propionato de clobetasol tópico. Tras 6 meses asintomático, se evaluó posible afectación sistémica, realizándose hemograma y bioquímica sanguínea (incluido proteinograma, velocidad de sedimentación globular [VSG], transaminasas, enzimas musculares y estudio lipídico), coagulación, inmunoglobulinas, complemento y analítica urinaria normales. Autoanticuerpos anti-ADN nativo, anti-SM, anti-Ro, anti-La, anti-Jo1, anti-scl y anticuerpo lúpico, inmunocomplejos y factor reumatoide negativos. ASLO, 693 U. Anticuerpos antinucleares (ANA) débilmente positivos con control posterior negativo. El estudio de poblaciones linfocitarias reveló un aumento del porcentaje de linfocitos B con disminución de los CD8. Serologías frente a hepatitis, virus de Epstein-Barr (VEB) y citomegalovirus negativos. Frotis faríngeo: positivo a Staphylococcus aureus. En el momento de esa consulta las lesiones cutáneas prácticamente habían desaparecido tras el tratamiento con corticoides tópicos.
Seis meses después ingresó en nuestro hospital por presentar una tumoración dura y dolorosa en muslo derecho de 15 días de evolución acompañada de dificultad para la deambulación y extensión incompleta con dolor a la palpación de la extremidad inferior derecha de 2 meses de evolución, que no mejoraba con ibuprofeno. Refería antecedente de traumatismo en esa zona con un tobogán, así como la aparición de manchas persistentes en zonas postraumáticas y ojeras oscuras coincidiendo con edema palpebral en una ocasión. En la exploración física destacaba la presencia de pápulas de Gottron sobre nudillos de IFP de segundo y quinto dedos de ambas manos y eritema en eminencia tenar y sobre los tendones flexores de cuarto y quinto dedos junto a extensión incompleta de extremidad inferior derecha (que producía cojera) y una zona dura e hiperalgésica palpable en muslo derecho, sin cambio de coloración y con aumento de 1 cm en el contorno del muslo. Hiperreflexia rotuliana bilateral con reflejos aquíleos normales. Signo de Gowers, negativo. No existía debilidad de la musculatura de la cintura escapular ni cervical.
Con la impresión diagnóstica de dermatomiositis juvenil se repitieron los exámenes complementarios previos con resultados similares. Los autoanticuerpos fueron negativos. El antígeno de histocompatibilidad (HLA) del paciente era A2, A32, B37, B44, CW5, CW7, BW4, DR11, DR13, DQ6, DQ7, DR52. La radiografía de extremidades inferiores mostró calcificaciones en el muslo derecho (fig. 1) y zonas de aumento de densidad de partes blandas no calcificadas en muslo izquierdo. La resonancia magnética (RM) puso de manifiesto alteración bilateral periférica de la señal en varios grupos musculares; en el lado derecho la afectación era más significativa a nivel proximal del semimembranoso y semitendinoso, con alteración difusa de la señal en su inserción proximal en la tuberosidad isquiática, lo que sugiere desinserción parcial; en el lado izquierdo la afectación era menos marcada y más distal y afectaba a los músculos grácil y sartorio y con menor intensidad a los aductores; también existía afectación de la inserción distal del músculo recto femoral (fig. 2). El electromiograma fue normal. Las exploraciones cardiológica y oftalmológica fueron normales. La biopsia confirmó el diagnóstico de dermatomiositis juvenil y se inició tratamiento con deflazacort (2 mg/kg/día), añadiéndose a los 2 meses metotrexato semanal (10 mg/m2 por vía subcutánea). La dificultad para la deambulación y la tumefacción mejoraron progresivamente, aunque no llegaron a desaparecer.
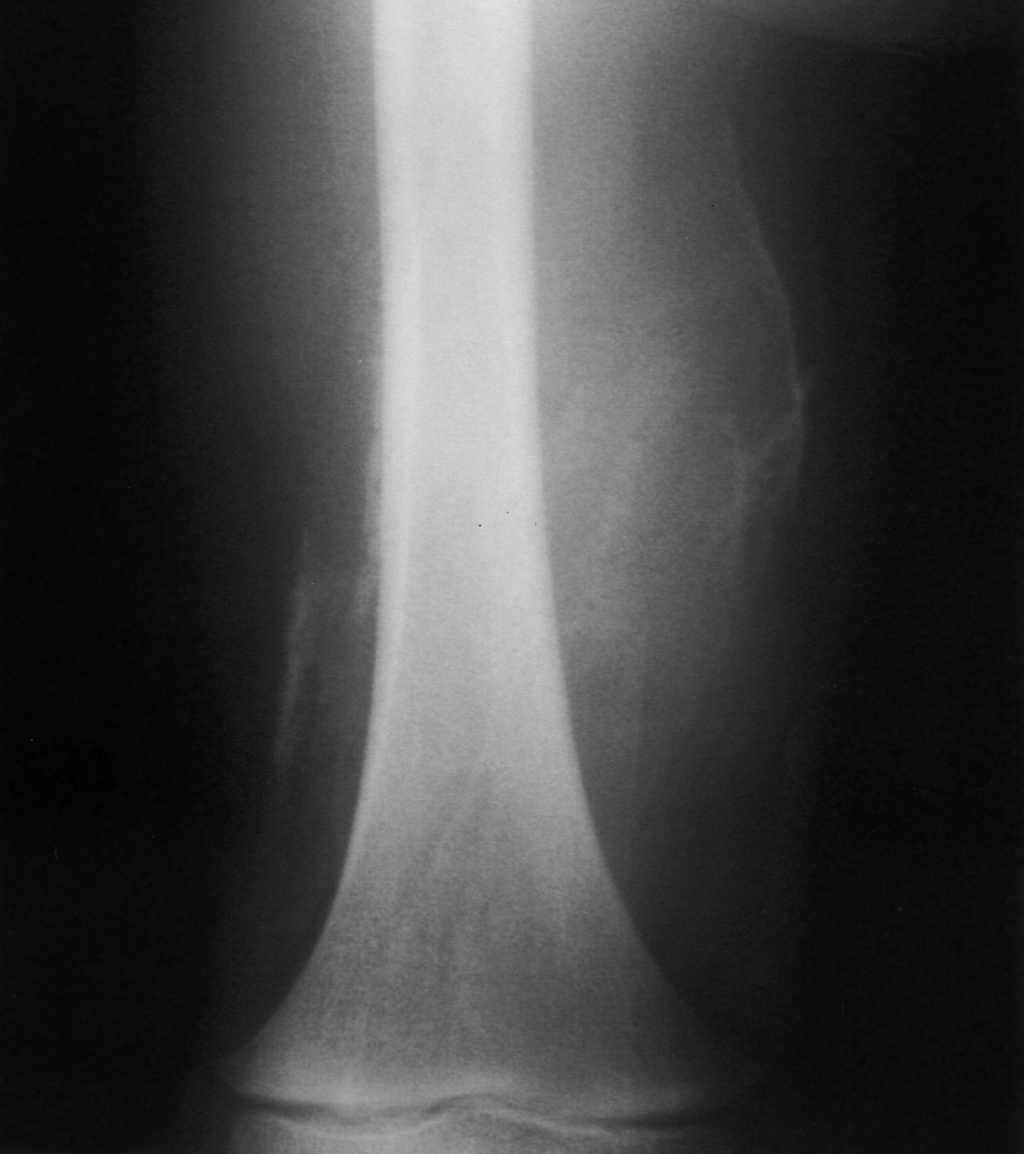
Figura 1. Radiografía muslo derecho: calcinosis.
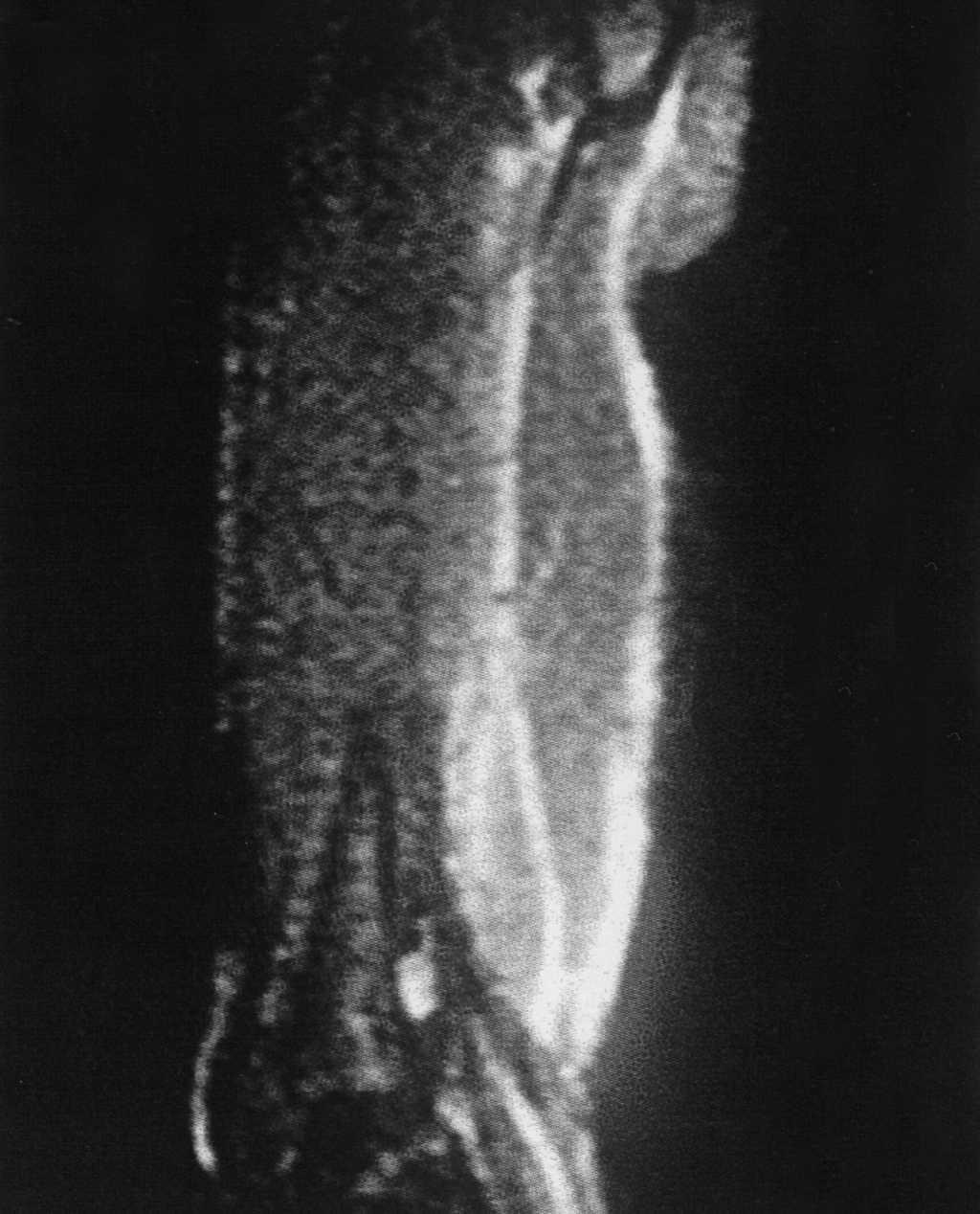
Figura 2.RM: alteración periférica (incluida afectación de la grasa de los septos intermusculares) de múltiples estructuras musculares fundamentalmente de grupos posteriores de ambos muslos, siendo más importante a nivel proximal de los músculos semitendinoso y semimembranoso con probable desinserción parcial de la tuberosidad isquiática y rotura fibrilar parcial periférica de los vientres musculares referidos.
Discusión
La dermatomiositis juvenil es una entidad poco frecuente en pediatría que debe sospecharse precozmente debido a sus implicaciones pronósticas y terapéuticas. La sintomatología se instaura generalmente de forma insidiosa durante 2-3 meses. Sólo en un tercio de los casos el inicio es agudo, y se relaciona con un peor pronóstico1. El síntoma fundamental es la debilidad muscular simétrica de predominio proximal acompañada de mialgia, hiperalgesia y edema indurado. Se afectan las cinturas escapular y pélvica y las musculaturas flexora del cuello, faríngea, hipofaríngea y velopalatina. El 75 % de los pacientes presenta síntomas cutáneos como primera manifestación (como en nuestro paciente) o en el curso de la enfermedad1. El signo inicial suele ser el edema periorbitario y cutáneo. Las lesiones patognomónicas son el eritema heliotropo y las pápulas de Gottron2. La fotosensibilidad puede manifestarse como eritema en cara anterior del tórax o palmar sobre los tendones flexores de los dedos. Otras manifestaciones cutáneas son las lesiones periungueales y las úlceras cutáneas1,3,4.
De las enzimas musculares, la creatinfosfoquinasa es la más específica y muestra niveles elevados en la fase inicial aguda y muy ligeramente alterados en la enfermedad de progresión lenta. En el 5-10 % de los pacientes la creatinfosfoquinasa es normal. La aldolasa se eleva de forma más significativa en los niños que en los adultos y es un indicador útil para el diagnóstico precoz, ya que su elevación precede a los síntomas musculares1,4. La electromiografía confirma el diagnóstico y muestra característicamente datos de miopatía y denervación, así como descargas repetitivas a alta frecuencia2. La biopsia muscular pone de manifiesto miositis inflamatoria con necrosis y fagocitosis de fibras musculares. No sería imprescindible su realización en niños con síntomas típicos1,2. En nuestro paciente sí fue necesaria para confirmar el diagnóstico dada la normalidad de las enzimas musculares y el electromiograma.
El tiempo transcurrido desde el primer síntoma hasta el diagnóstico oscila entre 2-6 meses. Las lesiones cutáneas suelen estar presentes y descritas en la primera evaluación de los pacientes, aunque no siempre contribuyen al diagnóstico precoz, como ocurrió en nuestro paciente. La calcinosis (habitualmente presente en los casos de curso grave2) aparece en la fase tardía de la enfermedad o en la remisión de la fase aguda, y es rara al inicio de la enfermedad. Puede tener múltiples localizaciones y generalmente es indolora, salvo las lesiones superficiales. Las lesiones periarticulares pueden dificultar la movilidad. El retraso en el diagnóstico y en el inicio del tratamiento de la dermatomiositis juvenil puede ser un factor que favorezca la aparición de calcinosis1,4. En la dermatomiositis amiopática el eritema es tratado habitualmente de forma tópica y posteriormente puede desarrollarse miositis con una incidencia más elevada de calcinosis.
Se observa una alta incidencia de infecciones estafilocócicas1 y un defecto quimiotáctico de los granulocitos en los niños con dermatomiositis juvenil que posteriormente desarrollarán calcinosis.
El diagnóstico diferencial se plantea con: miositis infecciosas, miopatías (distrofias musculares, miopatías metabólicas y endocrinas), enfermedades neurológicas (polineuritis, atrofias espinales, enfermedad de la unión neuromuscular), otras enfermedades del tejido conjuntivo (lupus eritematoso sistémico, esclerodermia, artritis idiopática juvenil, miositis por cuerpos de inclusión) y otras (medicamentosas, rabdomiólisis, tumores, miositis osificante progresiva)1. En nuestro caso, la presencia de calcinosis unida a la ausencia inicial de criterios diagnósticos que apoyaran la sospecha clínica de dermatomiositis juvenil nos hizo plantearnos otras causas de calcinosis. Descartada la etiología renal y dado el antecedente de traumatismo previo a la aparición de la tumoración nos planteamos la posibilidad de una miositis osificante traumática, también llamada osificación heterotópica. Esta entidad se caracteriza por la formación de hueso dentro del músculo como resultado de una lesión dentro de esta estructura. Los pacientes presentan una tumefacción sensible a la presión (evidente a los 15-20 días del traumatismo), dolor e impotencia funcional. Puede producirse por un único traumatismo o por múltiples traumatismos de pequeña intensidad. Las localizaciones más comunes incluyen el cuádriceps, lugar de asiento de la lesión de nuestro paciente. Por lo general, no necesita ningún tratamiento especial5.
En conclusión, aunque la dermatomiositis juvenil es una enfermedad poco frecuente, se debe sospechar ante la presencia de manifestaciones musculosqueléticas en un paciente con lesiones cutáneas previas.
