

La porfirias son un grupo de enfermedades de origen genético o adquirido causadas por una alteración en alguna de las enzimas que intervienen en la síntesis del grupo hemo1–5. Existen diversos tipos de porfiria según el lugar en que dicho proceso se ve interrumpido2,3,5–7 (fig. 1). Clínicamente, se diferencian las cutáneas y las de presentación aguda3,5, entre las cuales la porfiria aguda intermitente (PAI) es la más frecuente1,6,7. Se presenta con herencia autosómica dominante, con penetrancia incompleta y su causa reside en una mutación en el gen de la porfobilinógeno deaminasa (PBGD), localizado en el cromosoma 111.

Se presenta el caso de un recién nacido asintomático portador de una mutación causante de PAI, en el que se realizaron los estudios tras diagnosticar la enfermedad en la madre.
Gestante de 27 años con antecedentes de dolores abdominales recurrentes, vómitos y algias generalizadas, relacionados con el periodo premenstrual, catalogados de síndrome ansioso-depresivo. Se practica cesárea a las 31 semanas por corioamnionitis, con buena evolución del recién nacido. Tras el parto, la madre inicia náuseas, vómitos y mialgias asociados a hiponatremia grave. Con la orientación diagnóstica de secreción inadecuada de hormona antidiurética, se inician restricción hídrica y reposición de sodio sin mejoría del perfil hidroelectrolítico. En los días posteriores presenta un cuadro de convulsiones que requiere tratamiento con fenitoína. A las 48h del comienzo del tratamiento anticonvulsivo presenta parálisis de extremidades rápidamente progresiva, llegando hasta la tetraplejia. Recibe tratamiento con tiamina e inmunoglobulina humana sin respuesta, descartándose Beriberi y Guillain-Barre, respectivamente. Finalmente, tras varias hipótesis diagnósticas, se incluye porfiria en el diagnóstico diferencial. El estudio bioquímico confirma la sospecha diagnóstica, detectándose cifras elevadas de porfobilinógeno (PBG) y ácido delta-aminolevulínico (D-ALA) en orina, y un aumento importante de porfirinas en sangre. Se realiza el estudio genético que confirma la existencia de la mutación c.652del A en el intrón 11 del gen PBGD, responsable de PAI. Se administra tratamiento con hemina con mejoría clínica progresiva. Debido al carácter hereditario de la enfermedad, se realiza el cribado de la enfermedad en el niño. El estudio genético confirma la presencia de la mutación encontrada en la madre, siendo él portador heterocigoto de la enfermedad.
La porfiria se presenta habitualmente en adultos y aparece en forma de crisis agudas de síntomas gastrointestinales, neurológicos y psiquiátricos, con periodos libres de sintomatología1–3,5–8. Un 80-90% de los portadores de la mutación son asintomáticos durante toda su vida1,3,5,7,8. En niños, sólo se ha descrito la aparición de síntomas en los raros casos de homocigosis, en los que la enfermedad se manifiesta precozmente en forma de un proceso neurodegenerativo progresivo9.
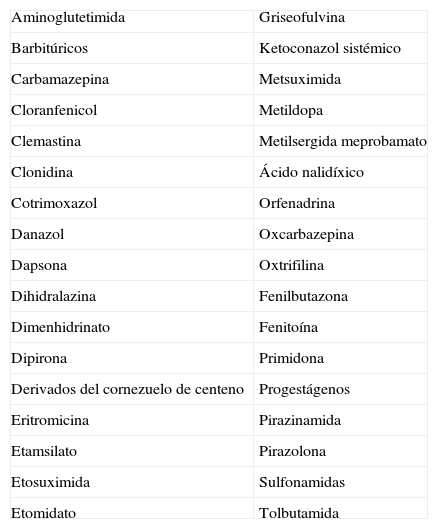
El déficit enzimático presente en las porfirias agudas predispone a los pacientes a los efectos de factores precipitantes que provocan un aumento de demanda de hemo hepático e inducen la síntesis de ALA sintasa, la primera enzima en la cadena del hemo5. Como consecuencia, se produce una acumulación de intermediarios en el hígado y el tejido nervioso, que presumiblemente son los responsables de la sintomatología. El alcohol, los procesos infecciosos, determinados cambios hormonales, el embarazo2,4,10, situaciones de ayuno o el consumo de determinados fármacos (tabla 1) son los factores desencadenantes más conocidos1,3,5–8.
Fármacos porfirógenos
| Aminoglutetimida | Griseofulvina |
| Barbitúricos | Ketoconazol sistémico |
| Carbamazepina | Metsuximida |
| Cloranfenicol | Metildopa |
| Clemastina | Metilsergida meprobamato |
| Clonidina | Ácido nalidíxico |
| Cotrimoxazol | Orfenadrina |
| Danazol | Oxcarbazepina |
| Dapsona | Oxtrifilina |
| Dihidralazina | Fenilbutazona |
| Dimenhidrinato | Fenitoína |
| Dipirona | Primidona |
| Derivados del cornezuelo de centeno | Progestágenos |
| Eritromicina | Pirazinamida |
| Etamsilato | Pirazolona |
| Etosuximida | Sulfonamidas |
| Etomidato | Tolbutamida |
Disponible en: www.orphan-europe.com.
El diagnóstico de una crisis aguda de porfiria se basa en la detección de precursores del hemo, fundamentalmente D-ALA y PBG, en orina1,3,5–7, cuyos niveles se encuentran elevados en todas las porfirias agudas en momentos de crisis porfírica, excepto en la porfiria ALA-deshidratasa, en que no existe aumento de PBG. La detección de porfirinas en orina, heces, eritrocitos o plasma es también útil para el diagnóstico y la orientación hacia el tipo de porfiria, ya que cada una de ellas tiene un patrón concreto de producción porfirínica. La confirmación definitiva de PAI se realiza mediante la determinación de la actividad de la PBG-D eritrocitaria1,3,5,6 y el análisis genético1,3,5.
La pieza fundamental en el manejo de la enfermedad es la prevención de las crisis, evitando factores desencadenantes6,7. No existe un tratamiento efectivo al 100% pero la infusión de hematina vía central tiene buenos resultados generalmente1,5–7. La administración de glucosa intravenosa u oral puede beneficiar a algunos pacientes1,5, así como el trasplante hepático en casos extremos1.
La detección de portadores asintomáticos de la mutación puede ayudar a reconocer y tratar precozmente las crisis, en muchas ocasiones infradiagnosticadas por su inespecificidad clínica. En estos pacientes un manejo exhaustivo de los posibles factores desencadenantes y el uso controlado de fármacos serán la pieza clave para el éxito en el control de la enfermedad.
Al Dr. Jordi To-Figueras y a la Dra. Cèlia Badenas, del Laboratorio de Bioquímica y Genética Molecular del Centro de Diagnóstico Biomédico (CDB) del Hospital Clínic de Barcelona, responsables del diagnóstico bioquímico y genético, respectivamente.
