


Introducción
El fenómeno de la inmigración en España ha acarreado la aparición, como problemas de salud pública, de patologías antaño muy poco comunes. Es el caso de la enfermedad de células falciformes (ECF) o drepanocitosis, término que engloba un conjunto de trastornos hereditarios con un núcleo común, la existencia de hemoglobina S (HbS) en proporción suficiente en un individuo como para producir manifestaciones clínicas. Esta HbS es el resultado de una alteración genética estructural de la cadena β de la globina debida a la sustitución de un único aminoácido con carga, el ácido glutámico, por otro aminoácido neutro, la valina.
La Sociedad Española de Hematología Pediátrica (SEHP) ha estado realizando desde el año 2000 la recogida de los pacientes afectados de drepanocitosis en toda España, pidiendo colaboración a los hospitales en relación con dicha sociedad, y recogiendo determinadas características de los mismos, tanto demográficas como las referidas a sus complicaciones clínicas y a las actuaciones diagnósticas y terapéuticas que en ellos se han llevado a cabo.
Tratamos de describir la experiencia del último año de recogida de datos (los datos recogidos en el año 2004 referidos a los pacientes en seguimiento en el año 2003), y reflexionar sobre la eficacia de la metodología de la recogida de datos y sobre determinados resultados. De esta forma tratamos de conocer de forma más precisa la incidencia de drepanocitosis en nuestro país para así intuir la magnitud del problema y colaborar mejor con el tratamiento y resolución de sus complicaciones.
Pacientes y métodos
La recogida de datos se ha realizado en el último año solicitando la información sobre los pacientes pediátricos con ECF, no sólo a los hospitales en relación con la SEHP, sino a todos los hospitales nacionales presentes en el Catálogo de Hospitales de la página web del Ministerio de Sanidad y Consumo, tanto a entidades públicas como privadas y dentro de éstas, a benéficas y no benéficas. En dicha web encontramos 775 hospitales. Un total de 467 recibieron nuestra petición de colaboración. El resto quedaron excluidos por ser hospitales sin camas pediátricas (hospitales traumatológicos, quirúrgicos, geriátricos, etc.).
Se han incluido en este registro a los pacientes de 0 a 18 años con ECF, y han quedado excluidos los portadores.
La información se solicitó de forma escrita a través de correo regular, durante noviembre y diciembre de 2003. En cada envío se introdujo una hoja de presentación del proyecto de recogida de datos y varias hojas de recogida tanto de datos iniciales como de seguimiento, según el protocolo de drepanocitosis de la SEHP del año 2002. Se fue recopilando la información enviada durante el primer trimestre de 2004.
Los datos recogidos en cada paciente fueron epidemiológicos, relativos a diagnóstico, tratamiento y complicaciones agudas y crónicas (tabla 1). La información se fue recogiendo en una base de datos, en el programa Microsoft Excel para Windows XP Professional, para su posterior análisis. En la descripción de resultados se han utilizado determinación de porcentajes, medias y rangos. Para ello, en este programa se creó una hoja en la que se fueron colocando en las columnas cada uno de los pacientes codificados con un número para preservar su privacidad, y en las filas, las variables en estudio mencionadas en la tabla 1. De cada variable se crearon varias categorías, o bien cuantitativas (como edad en meses), o bien cualitativas (como varón o mujer en sexo). Con la opción del programa Excel filtro y autofiltro se realizó el análisis descriptivo de cada paciente y cada variable. Los valores estadísticos mencionados se obtuvieron con las funciones que oferta dicho programa de suma, promedio, contar y max.

Resultados
Se recibió contestación, en un sentido positivo o negativo, de 40 hospitales, es decir, del 8,57 % de los hospitales a los que se solicitó información. Entre ellos, 25 hospitales tenían algún paciente aún en seguimiento. Se presentan agrupados según comunidades autónomas en la tabla 2. Además, otros hospitales, el Hospital de Cruces de Bilbao-Baracaldo y el Hospital General de Valencia, habían presentado pacientes pero se había perdido el seguimiento de los mismos. El Hospital de Olot (Girona) presentaba pacientes cuyo seguimiento se hacía en común con otros hospitales. Otros centros o no tenían pacientes con drepanocitosis o tenían sólo portadores (Complejo Hospitalario de Toledo, Hospital General de Valencia).
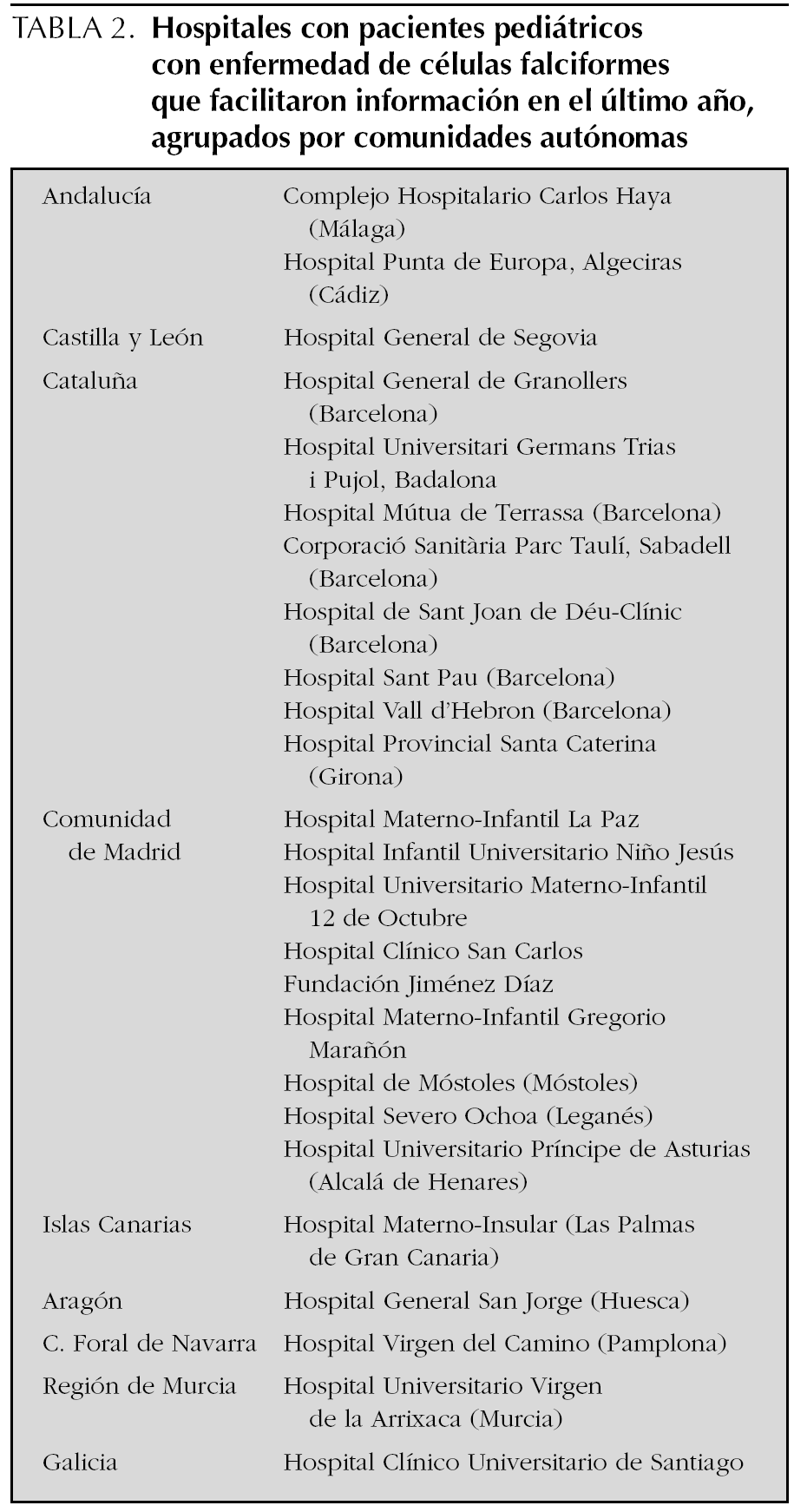
Un total de 6 hospitales mandaron pacientes al registro por primera vez. Fue el caso del Hospital Punta de Europa, Algeciras (Cádiz); el Hospital General de Segovia (Complejo Hospitalario de Segovia); el Hospital General de Granollers (Barcelona); el Hospital Provincial Santa Caterina (Girona); el Hospital Severo Ochoa (Leganés), y el Hospital General San Jorge (Huesca). El Hospital de Mataró manda fielmente cada año sus datos, pero no le fue posible en el último registro.
Se registraron un total de 223 niños. De ellos, 95 eran portadores y 138 eran enfermos homozigotos o doble heterozigotos. Del grupo de enfermos, 99 pacientes (72 %) continuaban aún en seguimiento (45 nuevos, incorporados al registro ese año) y en 39 pacientes se había perdido ya el seguimiento.
Respecto a los 138 enfermos, 68 (49 %) eran varones y 63 mujeres (46 %); en 7 pacientes (5 %) no obtuvimos este dato. De los 99 pacientes en seguimiento, 45 eran varones (45 %) frente a 48 mujeres (49 %); no se había referido sexo en 6 pacientes (6 %).
La media de edad en años de los pacientes en seguimiento fue 8,2, con un rango de 2 meses a 18 años. Del total de este registro, 10 habían sido diagnosticados por cribado neonatal (14 %).
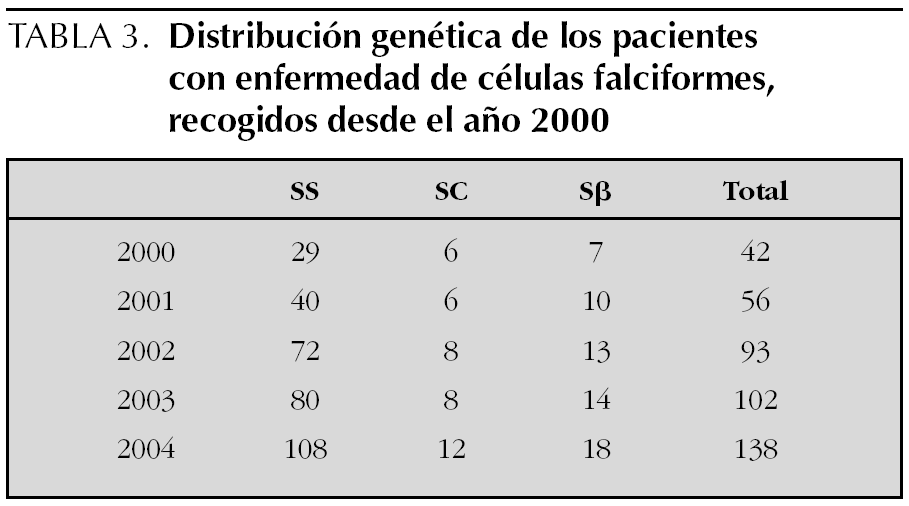
Los fenotipos de hemoglobina (Hb) fueron: Hb SS: 108 (78 %); Hb SC: 12 (9 %); Hb Sβ 0: 6 (4 %); Hb Sβ+: 12 (9 %). Sólo en 2 casos pudo confirmarse la consanguinidad de los padres. En la tabla 3 se muestra la distribución genética en los pacientes con enfermedad, desde el año 2000.
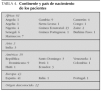
Respecto al continente y país de nacimiento, 61 pacientes (44 %) procedían de África; 3 (2 %), de Asia; 19 de América (14 %), y 43 (31 %) habían nacido en Europa. De estos últimos, casi el 100 % (41), habían nacido en España. En 12 casos (9 %) no pudo determinarse el origen (tabla 4).
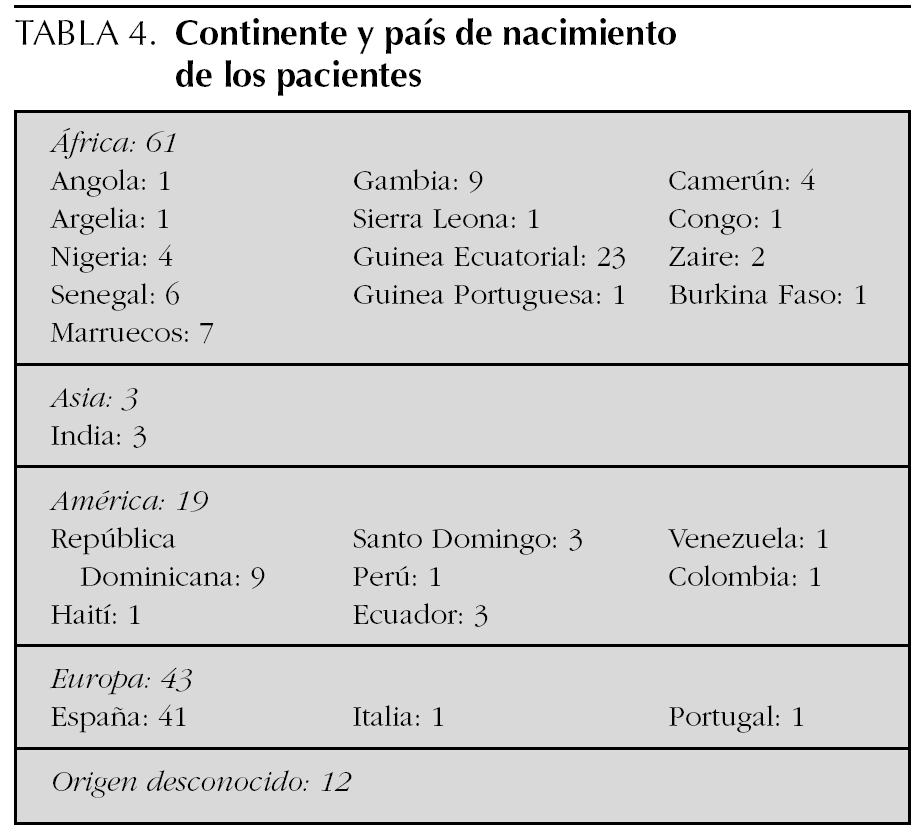
Recogimos el continente de origen del gen anómalo de los pacientes, es decir, el continente de origen de los padres, con los siguientes resultados: provenían de África 105 (76 %), de Asia tres (2 %), de América 20 (15 %) y de Europa dos (2 %). En 4 casos (5 %) no fue posible determinar el origen de los padres. De los niños nacidos en Europa (43), 39 parejas de padres eran de origen africano (91 %) y cuatro de origen americano (9 %).
En cuanto al seguimiento para prevención de cualquier tipo de accidente cerebrovascular agudo, se había realizado eco-Doppler transcraneal en 11 pacientes en el último año, en todos ellos con resultados dentro de la normalidad. Se llevó a cabo resonancia magnética (RM) cerebral en el último año en 22 pacientes, que fue normal en 17 pacientes (77 %) y patológica en cinco (6 %). Se había realizado exploración neuropsiquiátrica en 11 pacientes, en 7 de ellos (64 %) con resultado normal y en cuatro (36 %) con resultado patológico. Uno de los pacientes, un varón de 7 años que había sufrido accidentes isquémicos en años previos, presentaba imágenes de hiperintensidad en trayectos de la arteria cerebral anterior derecha, en el trayecto poscomunal y en cerebral media: padecía alteraciones motoras en pierna y pie izquierdos con un discreto déficit sensitivo en los dedos del pie izquierdo. Otro paciente varón, de 10 años, presentaba en la RM una pequeña imagen hiperintensa en sustancia ventricular alrededor de los atrios ventriculares; además, tenía problemas de aprendizaje. Un tercer varón, de 3 años, presentaba alteraciones en la arteria cerebral anterior bilateral en la RM y padecía un apreciable retraso psicomotor. La cuarta afectada era una joven de 15 años que, aunque presentaba en la RM una hipercaptación en la carótida interna derecha, estaba asintomática. La RM de la quinta paciente, de 5 años, mostraba una estenosis segmentaria destacable de la arteria carótida derecha intracraneal, menos intensa en igual zona en la carótida izquierda con estenosis leve en áreas supragenoideas de ambas carótidas; presenta frecuentes cefaleas y sensación de desvanecimiento frecuente al levantarse desde la posición horizontal.
El tratamiento seguido por los 99 pacientes aún en seguimiento fue sólo sintomático en 64 (65 %). Tuvimos constancia de que realizaron profilaxis con penicilina 21 de los 38 pacientes menores de 5 años (55 %) (en el resto no apareció el dato registrado). De los 61 pacientes mayores de 5 años, 21 (34 %) realizaban profilaxis con penicilina, 22 no lo hacían y en los restantes 18 pacientes mayores de 5 años no apareció el dato registrado. Fueron vacunados según calendario recomendado en el protocolo de la SEHP 60 pacientes (en el resto de pacientes no se ha registrado el dato).
Se administró terapia hipertransfusional en 3 pacientes, quelación por sobrecarga férrica en cuatro más y tratamiento con hidroxiurea en otros 27 pacientes. Respecto a estos últimos, la media de edad al inicio del tratamiento fue de 8,8 años, la HbF media (último valor obtenido durante el tratamiento) fue de 17,32 %; presentaron toxicidades transitorias 4 pacientes (15 %), hematológicas en todos los casos, sin que ninguno de ellos tuviera necesidad de supresión del tratamiento.
En el año 2004 no se registró ningún paciente que se hubiera sometido a trasplante de progenitores hematopoyéticos en el año anterior. De los datos recogidos de años previos, 3 pacientes del registro habían sido sometidos a esta terapia, de los cuales dos fallecieron y el tercero (trasplantado en el Hospital Vall D'Hebron de Barcelona) sigue vivo con buena evolución.
Un total de 14 pacientes habían recibido ese año transfusiones agudas de concentrado de hematíes.
Con respecto a la evolución de los 99 pacientes en seguimiento en el último año, presentaron complicaciones agudas (con necesidad de hospitalización) 21 pacientes: apareció dolor vasooclusivo en 14, síndrome torácico agudo en siete, hubo 6 casos de secuestro esplénico, fiebre sin foco en cinco y sepsis por Salmonella en un caso.
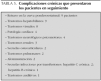
En el último año, 27 pacientes presentaron complicaciones crónicas (tabla 5).

Ese mismo año fallecieron 2 pacientes: uno por motivo desconocido (presumiblemente por problemas cardíacos) y el otro por causa infecciosa.
Discusión
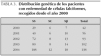
El fenómeno de la inmigración nos obliga a los pediatras a actualizarnos y familiarizarnos con problemas de salud que suponían hace años casos muy concretos. Así ocurre con la ECF o drepanocitosis, en sus diferentes formas clínicas. Se ha podido observar que ha ido aumentando la incidencia de pacientes pediátricos con esta enfermedad en los últimos años en España (tabla 3), de 42 pacientes a 138 pacientes, es decir, más del 200 % de enfermos en 5 años. Pero tal vez estas cifras podrían infravalorar la situación actual.
Por un lado, de la extrapolación teórica de una incidencia aproximada del gen drepanocítico en España por datos nacionales referidos a inmigración, esta cifra de 138 pacientes sería menor a la que podría esperarse. Los últimos datos del Padrón Municipal que aparecen en internet, en la dirección correspondiente al Instituto Nacional de Estadística, referidos al año 2003, revelan que en España existían entonces 107.402 inmigrantes procedentes del África subsahariana. No obstante, sería muy difícil determinar la frecuencia media del gen drepanocítico en el África subsahariana, parte del mundo en que es más alta, por ser muy variable según cada región concreta, incluso dentro de la zona denominada cinturón del paludismo1, donde se ha postulado que la mutación genética de la drepanocitosis proporciona una ventaja genética frente a la malaria 2. Además, recientemente se ha confirmado la ventaja de los portadores en este sentido 3. No obstante, es más frecuente en las zonas occidental, central y oriental del continente, donde se estima una incidencia de portadores del 5 al 40 % de la población, y es menos frecuente en las zonas norte y sur de África 4. De manera global, la prevalencia del gen es del 26 % en las zonas donde la malaria es endémica, y es más baja en otras regiones, por ejemplo, en Kenia, donde llega a mínimos del 7 % en algunos núcleos poblacionales 5, con la existencia de zonas de incidencia intermedia 6,7 y otras de alta incidencia al oeste del continente (Sierra Leona, Camerún, etc.) 8-10. Con todos estos datos podría extrapolarse la frecuencia media del gen drepanocítico en la población subsahariana podría ser del 14 % aproximadamente. Entonces, cabría esperar que unas 15.000 personas en España provenientes de este grupo portaran el gen. Según estos datos, podría calcularse que aproximadamente el 1-1,5 % de las personas que viven en el África subsahariana presentan ECF, lo que podría significar que 1.310 de estos inmigrantes fueran enfermos. Esto hace poco probable que sean sólo 138 pacientes pediátricos en España en estos momentos, aunque estas reflexiones hay que tomarlas con precaución, pues son extrapoladas de aproximaciones teóricas.
En segundo lugar, aunque los individuos descendientes del África subsahariana son los que presentan mayor riesgo de una alteración genotípica en relación con la HbS, sujetos procedentes del Mediterráneo, el Caribe, América Central y del Sur, Arabia 11 y el este de la India también presentan frecuencias no despreciables de esta alteración genética 12. Así, por ejemplo, en la India en conjunto existe una prevalencia de portadores de hasta el 34 % en algunas regiones en concreto, como es el caso de Madhya Pradesh, situada en la parte central de la misma 13. Por tanto, sería un error limitar a la raza negra esta gama de entidades nosológicas, ya que también están distribuidas entre otros núcleos poblacionales con otras razas 14.
Esto se une a que en los últimos años países donde la drepanocitosis no era prevalente anteriormente, comienza a ser un problema de salud pública, como es el caso de determinados países europeos como Irlanda 15, Noruega 16, Francia 17, Inglaterra 18, etc. Además, existen focos de la enfermedad en el sur de Italia, norte de Grecia y sur de Turquía. A todo esto habría que añadir un porcentaje de inmigrantes en España que por no estar legalizados, no han quedado registrados en el Instituto Nacional de Estadística.
Por tanto, teniendo en cuenta la incidencia teórica de portadores procedentes del África subsahariana, los genes que podrían provenir de otros continentes y que hay un porcentaje no estimable de inmigrantes ilegales en nuestro país, parecería probable la existencia de más niños con la enfermedad de los que hemos recogido.
Por otro lado, en cuanto al registro, llaman la atención otras dos cuestiones. Por un lado, sólo hemos recibido contestación del 8,57 % de hospitales a los que hemos pedido información. Pero además, llama la atención la escasa información que hemos obtenido de determinadas zonas geográficas o la nula información de algunas comunidades autónomas. Cabría pensar, por tanto, que existiría otro porcentaje de hospitales con pacientes, que no hayan podido enviar la información por falta de tiempo o por otros motivos. En un próximo año, para mejorar la adhesión de las distintas comunidades autónomas a la recogida de datos, se reforzará la estrategia con llamadas telefónicas a los principales centros de referencia. Este año se han adherido al registro 6 hospitales. Se intentará sumar otros nuevos en el próximo registro.
Nuestra distribución geográfica de pacientes se corresponde en frecuencias con lo descrito en la literatura médica, ya que un alto porcentaje de genes anómalos, el 76 %, provenían del África subsahariana, coincidiendo con zonas de elevada prevalencia de paludismo. Cabe destacar Guinea Ecuatorial como el país del que derivaban más pacientes.
Al ser la drepanocitosis un trastorno genético de herencia no ligado al sexo, no existiría teóricamente una mayor incidencia de un sexo sobre otro, como tampoco la apreciamos entre nuestros pacientes.
En cuanto a los distintos tipos genéticos de la enfermedad, en la muestra de nuestros 138 pacientes, el 78 % son homozigotos SS y el resto, dobles heterozigotos. Este porcentaje es parecido a los encontrados en otras localizaciones geográficas 19-21, aunque también hay que tener en cuenta que por ser la forma SS la más grave, es la que se diagnostica con más precocidad.
Se ha registrado la aparición de dolor vasooclusivo agudo con necesidad de hospitalización en 14 pacientes. La incidencia global de esta complicación, descrita como 0,4-1,0 episodios por paciente y año, es en realidad variable, pues depende de varios factores como la edad, el sexo del paciente, el tipo de enfermedad drepanocítica y del adiestramiento que el paciente tenga en el tratamiento de su dolor 22.
Los casos de fiebre sin foco que han precisado hospitalización han sido relativamente poco numerosos, el 5 % de los pacientes en seguimiento. La edad media de los pacientes que han padecido estos episodios es de 5,6 años, lo que coincide con estudios en series mayores que describen una edad media de 6,1 años, así como la coincidencia de estos episodios con otros de dolor vasooclusivo y síndrome torácico agudo, como ocurre en dos y uno de los pacientes de nuestra serie, respectivamente 23.
Una sepsis por Salmonella fue la causa del fallecimiento de uno de nuestros pacientes. Aunque la causa más frecuente de muerte en niños con ECF es la sepsis por Streptococcus pneumoniae24; también pueden producir el desenlace trágico otras infecciones.
Seis pacientes sufrieron secuestro esplénico. Aunque esta complicación ocurre en la mayoría de los casos en niños hasta los 5 años, se ha descrito incluso en adultos 25. De nuestros 6 casos, 1 de los que sufrió esta complicación tenía 15 años. Se comunicaron 7 casos de síndrome torácico agudo (el 7 % de los pacientes en seguimiento). En uno de los casos, coincidía con un secuestro esplénico. Se ha descrito en la literatura médica la aparición de síndrome torácico agudo hasta en el 20 % de los casos de secuestro esplénico 26.
Hasta en 27 de los pacientes en seguimiento (27 %) han aparecido complicaciones crónicas de diferente naturaleza. Las más frecuentes han sido los retrasos en la curva ponderoestatural y los trastornos hepatobiliares, seguidos de los oculares.
Un seguimiento adecuado de los pacientes con ECF puede prevenir en algunos casos y aliviar en otros las múltiples complicaciones susceptibles de aparición en esta enfermedad. La recogida de datos en todo el país nos lleva a ser conscientes de una realidad que progresivamente va adquiriendo importancia en el quehacer diario del profesional de la pediatría. Tenemos previsto continuar y mejorar esta recogida de datos para acercarnos cada vez más a la incidencia real de la ECF en España, poner experiencias en común entre los profesionales a partir de una detección de los problemas habituales de estos pacientes en nuestro entorno y conseguir un mejor tratamiento de esta patología en todos los aspectos asistenciales.
Agradecimientos
Damos las gracias a todos los miembros del Comité Elaborador del Protocolo Nacional de Drepanocitosis, a todos los hospitales que han tenido la amabilidad de contestarnos y a todas las personas que han hecho posible la recopilación de esta información, enviando los datos referentes a sus pacientes o a su hospital.
Correspondencia: Dra. M.B. García Arias.
Sección de Oncohematología Pediátrica.
Hospital Materno-Infantil Gregorio Marañón.
Máiquez, 9. 28009 Madrid. España.
Correo electrónico: martagarias@telefonica.net
Recibido en marzo de 2005.
Aceptado para su publicación en septiembre de 2005.
