




El síndrome linfoproliferativo autoinmune es la consecuencia de un defecto genético que compromete la apoptosis de los linfocitos. La linfoproliferación se manifiesta por adenomegalias y/o esplenomegalia crónica. El diagnóstico requiere demostrar el defecto de la apoptosis linfocitaria y el aumento de linfocitos T dobles negativos (TDN) que carecen de CD4 y CD8. Existe riesgo de desarrollar linfomas y enfermedades autoinmunes, sobre todo citopenias.
MétodosEstudiamos a un niño de 14 años con esplenomegalia de varios años de evolución con antecedentes familiares de esplenomegalia y adenomegalias. Se analiza el fenotipo de los linfocitos T y el defecto molecular del gen TNFRSF6 en el niño, su hermana y el padre. Se estudia el defecto de la apoptosis en el niño y su padre.
ResultadosEn el niño y su padre se confirman el defecto de la apoptosis de los linfocitos, el aumento de linfocitos dobles negativos (el 18 y 5%, respectivamente) y la misma mutación del gen TNFRSF6. La hermana presenta la misma mutación con un 16% de linfocitos doblemente negativos.
ComentariosLa esplenomegalia crónica familiar puede ser la única manifestación del síndrome linfoproliferativo autoimmune.
The autoimmune lymphoproliferative syndrome (ALPS) is caused by genetic defect in lymphocyte apoptosis. Chronic lymphadenopathy and splenomegaly are the consequence of lymphoproliferation. The diagnosis is based on the assessment of the defective lymphocyte apoptosis and the identification of lymphocyte T subset that are double negative (CD4-CD8-). The susceptibility to lymphoma and autoimmune diseases, mainly blood cytopenias is increased.
MethodsWe studied a 14 year-old boy with chronic splenomegaly and familial history of splenomegaly and lymphadenopathy. T lymphocyte phenotypes, and molecular defect of TNFRSF6 gene were studied in the child, his sister and his father. Lymphocyte apoptosis was also analysed in the child and his father.
ResultsThe boy and his father showed in vitro apoptosis defects, an increased number of double negative T lymphocytes (18% and 5%, respectively) and the same mutation in the TNFRSF6 gene. His sister had 16% of double negative T lymphocytes and the mutation in the TNFRSF6 gene.
CommentsChronic familial splenomegaly can be the only clinical sign of autoimmune lymphoproliferative syndrome.
El síndrome linfoproliferativo autoinmune (SALP) (OMIM 601859) es un proceso linfoproliferativo crónico causado por un defecto en la apoptosis de los linfocitos1. Fue descrito como síndrome de Canale-Smith en 1967 en dos niños con linfadenopatía, esplenomagalia, anemia y trombopenia2.
En 1995 se conoció el defecto molecular causado por una mutación del gen TNFRSF que codifica para la proteína Fas (CD95/APO1/TNFRSF6) que se encuentra en la membrana plasmática de los linfocitos3. Esta proteína interviene en la apoptosis de los linfocitos después de que han sido activados y han proliferado en respuesta al antígeno. La mutación de dicho gen es el defecto génico más frecuente, aunque se han descrito mutaciones de otros genes que codifican otras proteínas que intervienen en la apoptosis4,5,12 y casos sin mutación conocida en la actualidad.
El defecto en la apoptosis provoca un acúmulo de linfocitos en los órganos linfoides secundarios dando lugar a manifestaciones linfoproliferativas en forma de adenomegalias y esplenomegalia crónica6,7. Estos son los síntomas más frecuentes. Existe el riesgo de desarrollar enfermedades autoinmunes especialmente citopenias: anemia hemolítica autoinmune, trombopenia o neutropenia autoinmune6,7. Con menos frecuencia se ha descrito asociada a manifestaciones autoinmunes órganoespecíficas como glomerulonefritis8 y vasculitis cutáneas9. El pronóstico está en función del riesgo aumentado de desarrollo de linfomas, sobre todo del linfoma de Hodgkin10.
Es característica la acumulación tanto en sangre como en órganos linfoides secundarios de un subtipo de linfocitos T que no expresan la molécula CD4 ni CD8 (linfocitos T dobles negativos). La molécula que identifica a los linfocitos T es el receptor para el antígeno (TCR). Existen 2 tipos de TCR según tengan cadenas αβ o cadenas γδ. Los linfocitos TCR γδ suelen ser dobles negativos y pueden encontrarse en sangre periférica en un pequeño porcentaje (menor del 5% de los linfocitos). Los linfocitos TCR αβ expresan o bien CD4 o bien CD8. Por tanto, es anómalo el hallazgo de linfocitos T TCR αβ que no expresen ni CD4 ni CD8. Esta subpoblación linfocitaria es la que está elevada en este síndrome. También es frecuente la hipergammaglobulinemia.
Para confirmar el diagnóstico es preciso demostrar in vitro un defecto de la apoptosis de los linfocitos11.
Los criterios diagnósticos son 3: 1. síntomas clínicos de linfoproliferación; 2. linfocitos T dobles negativos (CD3+αβ+CD4-CD8-) en número superior al 1% de los linfocitos totales; 3. demostración in vitro del defecto de la apoptosis de linfocitos. Apoyan el diagnóstico la existencia de una enfermedad autoinmune, antecedentes familiares de SALP, el hallazgo de linfocitos dobles negativos en la biopsia del ganglio linfático o del bazo, la hipergammaglobulinemia y la mutación del gen que codifica la proteína FAS (tabla 1).
Síndrome linfoproliferativo autoinmune. Criterios diagnósticos
|
Apoyan el diagnóstico
|
Niño de 14 años controlado por esplenomegalia desde los 4 años de edad.
Antecedentes familiares: hermana de 12 años, recientemente diagnosticada de esplenomegalia. Padre con esplenomegalia y antecedentes de adenomegalias que han precisado exéresis. Abuelo y tía paterna con antecedentes de adenomegalias. No se registran datos de enfermedades autoinmunes ni linfomas.
Antecedentes personales: presentó adenomegalias generalizadas a los 2 años de vida y varios episodios de urticaria aguda.
Exploraciones complementarias:
Hemograma: Hg 13,8g/dl; Leucocitos 5.900/μl; linfocitos 1931/μl; Neutrofilos 2.800/μl; plaquetas 179.000/μl.
Bioquímica: proteínas totales 8,6mg/dl.
Inmunoglobulinas: G 2440mg/dl; Ig A 234mg/dl; Ig M 221mg/dl; Ig E 2290KU/l.
ANA y autoanticuerpos (factor reumatoide, anticuerpos antitiroideos, anticardiolipina, antitransglutaminasa, antimitocondrial y antimúsculo liso) negativos.
Ecografía: gran esplenomagalia de 18cm y adenopatías de hasta 8–9mms de diámetro en FID.
Estudio inmunológico:
Estudio de poblaciones linfocitarias mediante citometría de flujo:
- •
Linfocitos T totales: 74%=1.429/μl.
- •
Linfocitos T α/β+: 61%=1.178/μl.
- ○
Linfocitos Tα/β+CD3+CD4+: 31%=599/μl.
- ○
Linfocitos Tα/β+CD3+ CD8+: 29%=560/μl.
- ○
Linfocitos T dobles negativos (α/β+CD4 −CD8 −): 18%.
- ○
Estudio funcional de la apoptosis: Defecto en la apoptosis en el niño y su padre.
Para el estudio funcional de la apoptosis se estimulan los linfocitos durante 8 días con PHA+IL2, posteriormente se induce la apoptosis linfocitaria utilizando el anticuerpo monoclonal anti-CD95 y se cuantifica por citometría de flujo mediante la tinción con anexina V/Ioduro de Propidio12,17.
Estudio genético del gen TNFRSF6 que codifica para la proteína Fas12,17: Delección en heterozigosis de una guanina en el nucleótido 774 codón 178. Esta alteración provoca la aparición de un codon stop temprano en la posición 199. Esta delección no estaba previamente publicada en la literatura.
Estudio familiarHermana de 12 años:
Hemograma: Hg: 12,5gr/dl; leucocitos 5.300/μl; neutrófilos: 2900/μl; plaquetas: 158.000/μl.
Bioquímica: proteínas totales: 7,2mg/dl.
Inmunoglobulinas: Ig G: 1810mg/dl; Ig A: 263mg/dl; Ig M: 73,8mg/dl.
ANA y autoanticuerpos negativos
Ecografía: esplenomegalia de 16cm. Adenopatias en FID de hasta 8mm de diámetro.
Estudio Inmunológico:
Linfocitos totales 1214 células/μl.
- •
Linfocitos T totales: 62%=753/μl.
- •
Linfocitos T α/β+: 49%=595/μl.
- ○
Linfocitos T α/β+CD3+CD4+: 33%=401/μl.
- ○
Linfocitos T α/β+CD3+CD8+: 18%=219/μl.
- ○
- •
Linfocitos T dobles negativos (α/β+CD4 −CD8 −): 16%.
Se confirma la misma mutación del gen TNFRSF6.
Padre: Se encuentra un aumento de linfocitos T dobles negativos con un valor del 5% del total de linfocitos y un defecto del estudio funcional de la apoptosis. Es portador de la mutación descrita anteriormente en el gen TNFRSF6 (fig. 1).
Discusión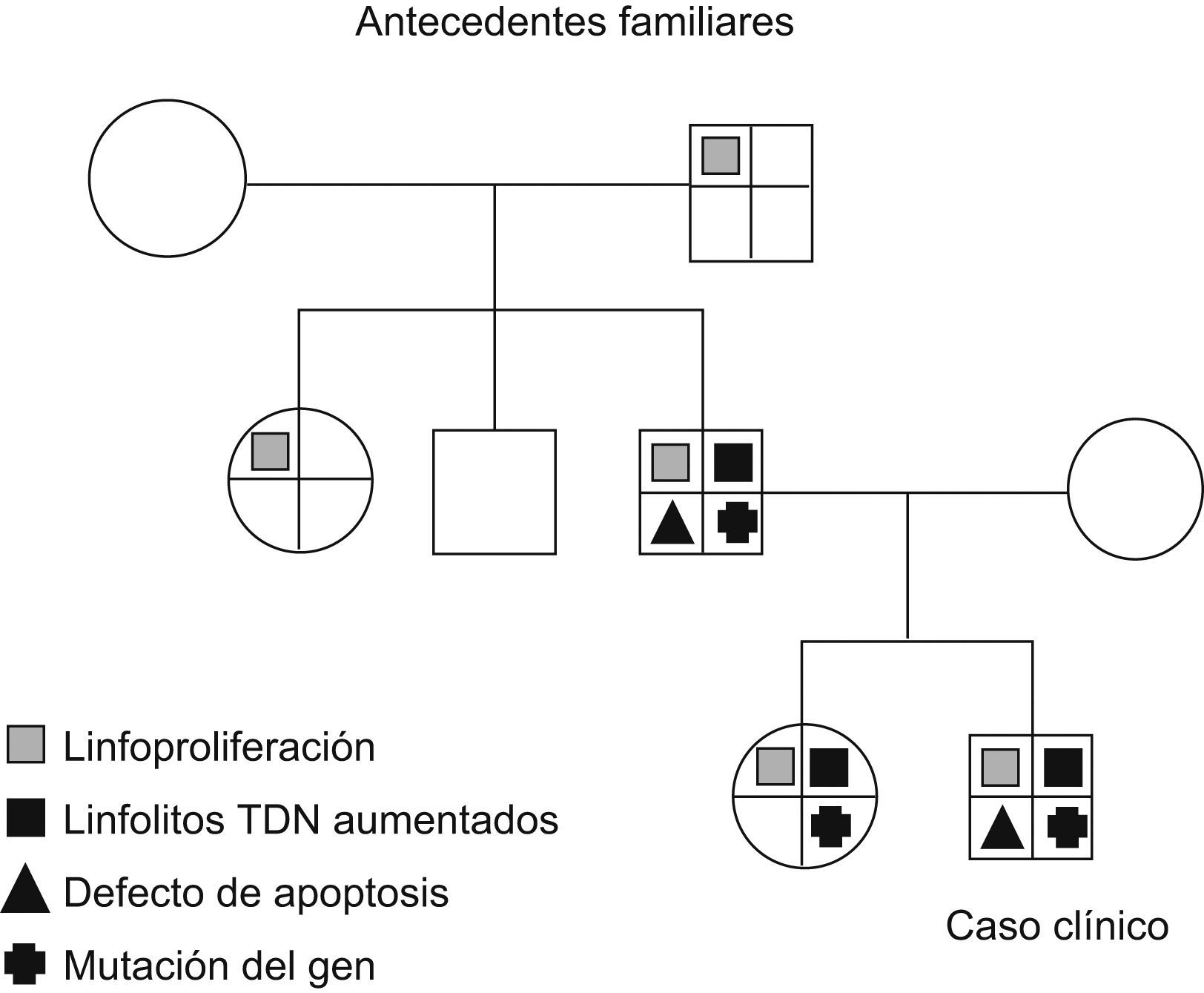
El síndrome linfoproliferativo autoinmune es una entidad reconocida recientemente.
Se considera una inmunodeficiencia primaria por defecto en la regulación inmune13. En la última clasificación de las inmunodeficiencias primarias se incluyen 8 grupos de enfermedades que cursan con defectos de la inmunidad, siendo uno de ellos los que cursan con defectos en la regulación inmune (tabla 2).
Inmunodeficiencias primarias. Clasificación
|
El síndrome linfoproliferativo autoinmune junto con el IPEX (disregulación inmune asociada a poliendocrinopatía y enteropatía autoinmune) y poliendocrinopatía, candidiasis y distrofia ectodérmica (APECED) constituyen 3 síndromes incluidos en el grupo de defectos en la regulación inmune cuyos defectos genéticos se asocian al desarrollo de enfermedades autoinmunes.
Existen 8 casos registrados en el Registro Español de Inmunodeficiencias Primarias14 (REIDP). En EE.UU. hay más de 250 casos registrados en el National Human Genome Research Institute15 con datos clínicos y analíticos de 62 pacientes. La esplenomegalia es el hallazgo más constante, se encuentra en el 90% de los pacientes. Le sigue en frecuencia la adenomegalia en el 87% de los casos. Dentro de las manifestaciones autoinmunes, el 53% presentaba anemia hemolítica autoinmune, el 13% neutropenia y otro 13% trombopenia. Dos de los 62 casos desarrollaron linfoma15.
En España se han publicado tres niños con el síndrome linfoproliferativo autoinmune con mutaciones en el gen TNFRSF616,17. Un niño de 5 años con esplenomegalia desde los 15 meses y antecedentes familiares de anemia hemolítica autoinmune y una niña controlada por esplenomegalia y adenopatías desde los 4 años que desarrolló un linfoma a los 11 años. El tercer caso es un varón con los criterios típicos de síndrome linfoproliferativo autoinmune. Además, en 2006 se publicó el caso de una paciente con síndrome linfoproliferativo autoinmune con una mutación en homozigosis en el gen TNFSF6 que codifica para FasLigando12 y que también cumplía los criterios típicos del síndrome linfoproliferativo autoinmune.
En nuestro caso, la sospecha clínica se planteó ante el hallazgo de esplenomegalia en la hermana junto con los antecedentes familiares.
Creemos justificado descartar este síndrome en los casos de esplenomegalia crónica no explicada, citopenias crónicas o linfomas sobre todo si existen antecedentes familiares.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
