





Introducción
La mucopolisacaridosis tipo I (MPS-I) es una enfermedad metabólica lisosomal debida a un déficit enzimático de α -L-iduronidasa, que se traduce en una acumulación de glucosaminoglucanos (GAG) dermatán y heparán sulfato en órganos y tejidos y en un aumento de su excreción urinaria. Los GAG son el resultado de la proteólisis de los proteoglucanos que constituyen la matriz extracelular. La amplia distribución del material de depósito da origen a una sintomatología progresiva y multisistémica.
La MPS-I es una entidad poco frecuente, con una incidencia de aproximadamente uno de cada 100.000 recién nacidos vivos y una prevalencia de uno por cada 400.000 habitantes 1. Se hereda de forma autosómica recesiva y hay descritas más de 80 mutaciones diferentes en el gen IDUA (locus 4p16.3). La gran variabilidad alélica se traduce en una elevada heterogeneidad fenotípica cuya gravedad depende de la actividad enzimática existente y del genotipo de cada paciente. Dentro de la MPS-I, el síndrome de Schie es la forma menos grave, el síndrome de Hurler-Schie es la forma intermedia y el síndrome de Hurler la de mayor gravedad, con mortalidad por lo general por causa cardiorrespiratoria en la primera década de la vida.
A pesar de esta gran heterogeneidad genotípica y fenotípica, existen dos mutaciones que son las más frecuentes en la población caucásica europea, la W402X y la Q70X, mutaciones sin sentido que dan origen a la formación de un codón "stop" prematuro y como consecuencia a una proteína truncada inactiva. Ambas mutaciones, si se heredan en homocigosis o heterocigosis combinada, originan formas graves de la enfermedad, con inicio precoz y muerte temprana 2-5.
El diagnóstico definitivo de la enfermedad requiere la cuantificación de GAG en orina y un estudio enzimático específico en piel (fibroblastos) y/o sangre (leucocitos, suero o plasma), que demuestre una actividad enzimática existente menor al 1 % de la actividad normal. El diagnóstico prenatal (en vellosidades coriales y/o amniocitos) y la detección de portadores (por análisis molecular) son posibles en la actualidad 6,7.
Se comunican 2 casos de MPS-I (síndrome de Hurler) en tratamiento con enzima recombinante humana α -L-iduronidasa, con el objetivo de analizar su eficacia y seguridad.
Observación clínica
Estudio prospectivo de 2 casos de MPS-I (síndrome de Hurler) de 4,8 años y 17 meses de edad (al inicio de la intervención) en tratamiento con enzima recombinante humana α -L-iduronidasa durante 52 y 28 semanas, respectivamente (tabla 1). se administraron 100 U/kg/semana por vía intravenosa de enzima recombinante humana diluida en suero fisiológico durante 4 h y se evaluó la eficacia y seguridad del tratamiento durante el tiempo que duró el estudio. De forma profiláctica se administraron antihistamínicos sedantes y antipiréticos orales, previamente a la dosis enzimática correspondiente.
Para evaluar la eficacia y la seguridad en el caso 1 (varón de 4,8 años, homocigoto W402X) y en el caso 2 (mujer de 17 meses, heterocigota para W402X) se realizaron determinaciones analíticas habituales con perfil hepático, lipídico y de coagulación, electrocardiograma, ecocardiograma, poligrafía cardiorrespiratoria durante el sueño; índice apnea-hipopnea/hora, electromiograma, ecografía abdominal, serie ósea, examen neurológico, otorrinolaringológico y auditivos seriados (incluyendo potenciales auditivos evocados) y se valoró clínicamente la movilidad articular, deambulación, la expresión y el fenotipo facial, de manera periódica. Así mismo, en ambos casos se realizaron controles analíticos seriados de los GAG urinarios mediante la prueba de metacromasia de Berry y espectrofotométrica con el colorante 1-9 dimetil metileno azul (DMB).
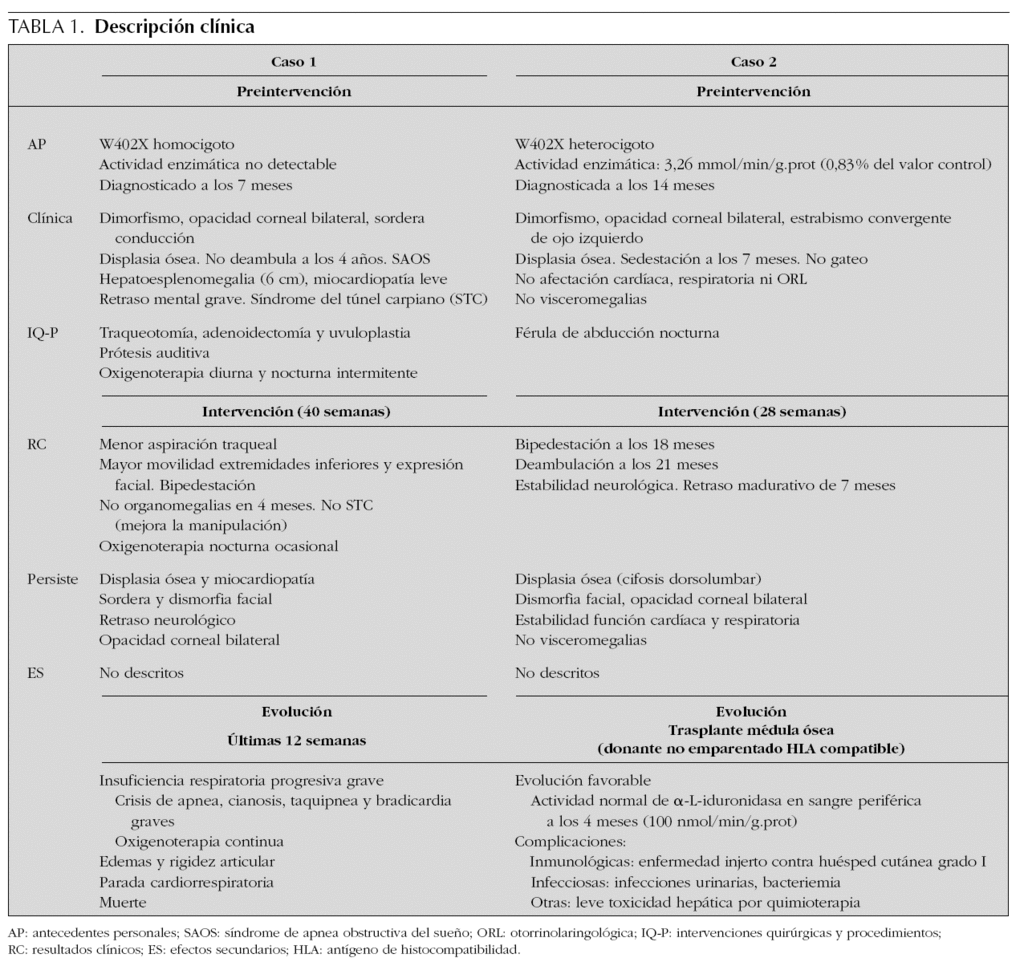
En el caso 1, tras 40 semanas de intervención, se observó una mejoría clínica de la insuficiencia respiratoria crónica, que se tradujo en una menor aspiración de secreciones traqueales con requerimientos de oxigenoterapia nocturna ocasional. También mejoró la expresión facial (sonrisa social), la manipulación manual (desaparición del síndrome del túnel carpiano) y la movilidad articular de extremidades inferiores posibilitando la bipedestación ocasional con apoyo. La organomegalia desapareció a los 4 meses de la intervención. La displasia ósea y el retraso neurológico grave persistieron a pesar del tratamiento enzimático sustitutivo. No se produjeron modificaciones significativas de la miocardiopatía (crecimiento del ventrículo izquierdo no obstructivo), las opacidades corneales bilaterales, la sordera de conducción ni la dismorfia facial. La mejoría clínica producida durante las primeras 40 semanas de la intervención coincidió con una disminución marcada de los GAG en orina (comprendida entre el 59,6 y el 72,13 %). Sin embargo, durante las últimas 12 semanas se produjo una agudización de la insuficiencia respiratoria crónica con episodios de apnea, cianosis y bradicardia frecuentes que precisaron hospitalización en la unidad de cuidados intensivos y finalmente se produjo la muerte en situación de insuficiencia ventilatoria aguda con hipoxemia marcada. Analíticamente se tradujo en un discreto incremento (22,5 %) de las concentraciones urinarias de GAG en la semana 48 de intervención con un estancamiento final posterior (tabla 1) (fig. 1).
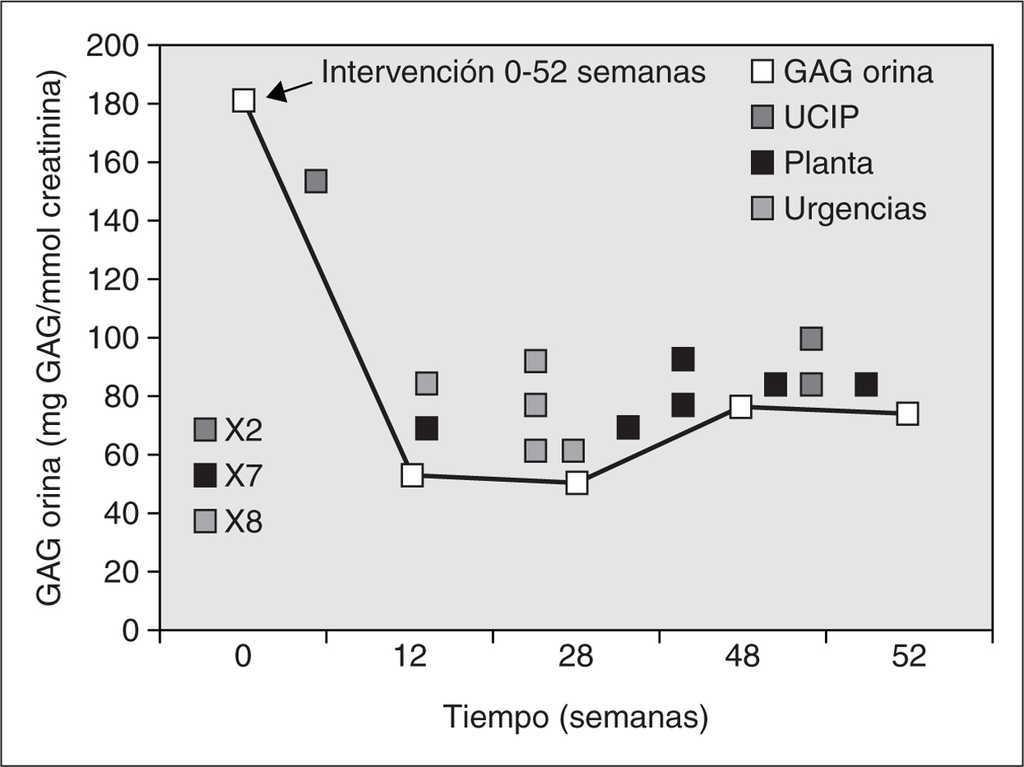
Figura 1. Monitorización de glucosaminoglucanos (GAG) en orina y su correlación clínica durante el tratamiento enzimático sustitutivo con α-L-iduronidasa durante 52 semanas en el caso 1. Antes de la intervención requirió un gran número de hospitalizaciones. Durante las primeras 35-40 semanas de tratamiento disminuyeron notablemente los GAG en orina y permaneció clínicamente estable. En las últimas 12 semanas aumentaron discretamente los GAG urinarios coincidiendo con una desestabilización clínica grave. Finalmente, el paciente falleció por fallo respiratorio agudo.
En el caso 2, tras 28 semanas de tratamiento enzimático sustitutivo, se observó una estabilización del desarrollo neurológico con un retraso madurativo de aproximadamente 7 meses. La bipedestación se produjo a los 18 meses y la deambulación a los 21. No se constataron modificaciones significativas en la displasia ósea (cifosis dorsolumbar marcada), la dismorfia facial ni en las opacidades corneales bilaterales y continuaron estables las funciones cardíaca y respiratoria, precisando ingresos hospitalarios únicamente por procesos intercurrentes. No se encontraron organomegalias. Analíticamente se cuantificó periódicamente la eliminación urinaria de los GAG con un descenso marcado (52,68 %) de los mismos en la semana 28 de intervención. Posteriormente, al disponer de un donante compatible, se realizó un trasplante alogénico de médula ósea (TMO) de donante no emparentado (Hospital Vall d'Hebron, Barcelona). Las concentraciones y la actividad de α -L-iduronidasa en sangre periférica fueron normales 4 meses después del trasplante. Como complicaciones presentó una enfermedad de injerto contra huésped cutánea grado I que se resolvió con corticoterapia, y procesos infecciosos intercurrentes que se resolvieron con antibioterapia intravenosa (tabla 1) (fig. 2).
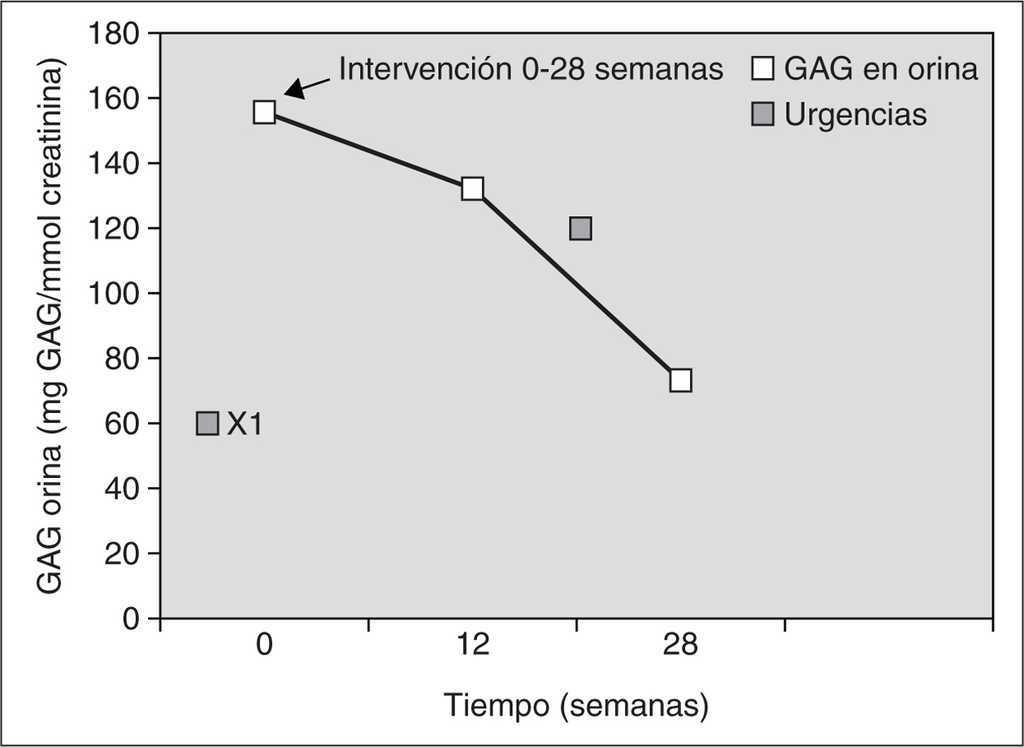
Figura 2. Monitorización de glucosaminoglucanos (GAG) en orina y su correlación clínica durante el tratamiento enzimático sustitutivo con α-L-iduronidasa durante 28 semanas en el caso 2. Previamente a la intervención no requirió ningún ingreso. Desde el inicio del tratamiento hasta el final se produjo una notable disminución paulatina de los GAG urinarios. Clínicamente permaneció estable sin adquisición de factores regresivos.
No se produjeron efectos secundarios significativos en ninguno de los dos casos, a excepción de un episodio aislado de hipertermia durante la infusión del tratamiento enzimático sustitutivo en el caso 2.
Discusión
En 1964 De Duve propuso por primera vez el tratamiento enzimático sustitutivo como opción terapéutica en las enfermedades de depósito lisosomal 8. Posteriormente, en 1981, Hobbs et al 9 observaron una mejoría clínica y analítica en un lactante afectado de MPS-I (síndrome de Hurler) tras trasplante de médula ósea de donante emparentado. Actualmente, ambas opciones terapéuticas continúan siendo los principales pilares del tratamiento de estos pacientes. El TMO y de células madre hematopoyéticas ofrecen la posibilidad de estabilizar el deterioro neurológico progresivo de estos pacientes. La terapia génica constituirá probablemente el tratamiento del futuro.
En enero de 2001 se publicó un ensayo clínico (fase I y II) realizado en 10 pacientes afectados de MPS-I, para evaluar los efectos clínicos y analíticos del tratamiento enzimático sustitutivo 10. Tres años después (mayo de 2004) se comunicaron los resultados de un ensayo clínico (actualmente en fase III) multicéntrico, internacional, aleatorizado, doble ciego, realizado en 45 pacientes afectados de MPS-I, que analizaba la eficacia y seguridad de esta opción terapéutica 11. Se constata una mejoría notable de la calidad de vida, con un incremento de la capacidad física (incremento de 38,1 m en el walking test) y de la función respiratoria (aumento de la capacidad vital forzada del 11,3 % y disminución del índice de apneas e hipopneas del sueño de 11,4 episodios por hora). Así mismo, se produce una disminución del tamaño de las visceromegalias (cuantificado por RM) con una normalización de la hepatomegalia en el 73 % de los sujetos estudiados.
A pesar del aumento de la movilidad articular por disminución de la acumulación del material de depósito en las partes blandas, la displasia ósea característica de estos pacientes no mejora, debido al alto peso molecular de la α -L-iduronidasa que dificulta el paso a través de la matriz intercelular del cartílago y el hueso. Así mismo, el paso a través de la barrera hematoencefálica, si se administra de forma intravenosa, también resulta difícil y, por tanto, puede no ser capaz de detener el deterioro neurológico grave característico del síndrome de Hurler. Sin embargo, se ha descrito una reducción significativa de los GAG en meninges y tejidos cerebrales tras la administración intratecal de α -L-iduronidasa en perros afectados de MPS-I, ofreciendo por tanto, una opción terapéutica novedosa y efectiva de esta entidad y de otras enfermedades de depósito lisosomal que cursen con deterioro neurológico intenso. Analíticamente se produce una disminución marcada de los GAG en orina, con una reducción media del 54,1 % a las 26 semanas de la intervención 12-14.
Se han comunicado dos tipos de efectos secundarios relacionados con el tratamiento enzimático sustitutivo: fenómenos de hipersensibilidad transitoria relacionados con la infusión (fiebre, exantema cutáneo, cefalea, hipertermia y urticaria), y la presencia de anticuerpos contra la enzima recombinante humana α -L-iduronidasa.
Las reacciones de hipersensibilidad se presentan habitualmente (48 %). Suelen ser fácilmente controlables disminuyendo la velocidad de la infusión y administrando fármacos inmunoprofilácticos (antes o durante la infusión) y por lo general no requieren la interrupción del tratamiento.
Se ha descrito la presencia de inmunoglobulina G anti-α -L-iduronidasa en prácticamente la totalidad de los pacientes tratados (40-91 % con seroconversión a los 52,6 ± 24,1 días). También se ha observado una disminución de su reactividad tras 6 meses de tratamiento, lo que sugiere el desarrollo de tolerancia inmunológica frente a la enzima recombinante humana. La disminución de la reactividad inmunitaria parece explicar el mínimo impacto que los anticuerpos ejercen en la eficacia del tratamiento enzimático a lo largo del tiempo 15.
A pesar de ello, el 83 % de los pacientes incluidos en el estudio multicéntrico internacional comentado anteriormente 11 eran formas intermedias de MPS-I y, dado que los 2 casos que se han estudiado eran graves (MPS-I, síndrome de Hurler), quizá los resultados de este estudio no fueran totalmente extrapolables a nuestros pacientes. Sin embargo, los resultados clínicos y analíticos obtenidos en un estudio realizado en 20 pacientes menores de 5 años afectados de síndrome de Hurler en tratamiento enzimático sustitutivo (actualmente en fase II) han sido similares a los comentados anteriormente 16. Se produjo una reducción de los GAG en orina del 57,05 % en la semana 26 de tratamiento y en general fue bien tolerado (25 % de hipersensibilidad transitoria). Los valores de GAG en orina obtenidos a lo largo de la intervención fueron mayores que en los pacientes afectados de formas intermedias, quizá porque la mayor cantidad de material de depósito lisosomal existente en los niños con síndrome de Hurler, requiera mayor tiempo o mayor dosis enzimática para corregirse.
En nuestros 2 casos, la terapia enzimática sustitutiva con enzima recombinante humana α -L-iduronidasa ha sido efectiva. En el caso 1, la intervención fue planteada con fines paliativos desde el comienzo debido a su estado clínico muy evolucionado. Durante las primeras 40 semanas de intervención, se produjo una mejoría tanto clínica como analítica con estabilización del cuadro clínico progresivo que presentaba, mejorando notablemente su calidad de vida. Finalmente, tras 52 semanas de intervención y debido a su enfermedad respiratoria de base, se produjo la muerte por insuficiencia respiratoria aguda. En el caso 2, la intervención se realizó de forma precoz y se observó una estabilización clínica tras 28 semanas de tratamiento enzimático sustitutivo sin la adquisición de factores regresivos neurológicos, respiratorios ni cardíacos. Se ha realizado con éxito un trasplante de médula ósea de donante no emparentado, con actividad enzimática normal en sangre periférica a los 4 meses del trasplante.
Para valorar la seguridad, además de los exámenes clínicos y analíticos periódicos, se han estudiado las concentraciones de IgG anti-α -L-iduronidasa cada 4 semanas, resultados pendientes en la actualidad.
A pesar de los beneficios que aporta la terapia enzimática sustitutiva, el TMO y de células madre hematopoyéticas incrementan la supervivencia y detienen o incluso mejoran la función cognitiva de estos pacientes. El trasplante se indica siempre que no exista un deterioro neurológico grave ya establecido y que el cociente intelectual (CI) sea superior a 70. Los resultados obtenidos mejoran cuando la edad es inferior a los 2 años, debido al beneficio notable del crecimiento de dendritas y conexiones axonales que persisten hasta aproximadamente los 3 años de edad. Tras la realización del TMO se consigue la corrección metabólica mediante la transferencia de la α -L-iduronidasa procedente del donante a los lisosomas del receptor, con la consiguiente filtración de los GAG circulantes e hidrólisis posterior. Se ha descrito una reducción marcada de las visceromegalias, de los episodios obstructivos de las vías respiratorias superiores y una mejoría notable de la dismorfia facial y de la movilidad articular. A pesar de la existencia de dificultades de aprendizaje en la mayoría de los pacientes tras 10 años de seguimiento, la función neurocognitiva de estos permanece estable, sin que se observe retraso mental grave en ninguno de ellos con CI en el rango de la normalidad (75-103). Sin embargo, las opacidades corneales no presentan modificaciones significativas y debido al elevado peso molecular de la α -L-iduronidasa, la disostosis múltiple característica de estos pacientes persiste a pesar del tratamiento 17-20.
Además de los efectos secundarios derivados de la inmunodepresión pre y postrasplante y del posible rechazo de este, cabe destacar la hemorragia pulmonar como complicación poco habitual, pero a tener en cuenta por la gravedad que conlleva 21.
Actualmente, ante la carencia de donantes de médula ósea histocompatibles se está realizando trasplante de células madre hematopoyéticas procedentes de cordón o de sangre periférica con resultados satisfactorios. Tras la mieloproliferación y maduración de las células inmaduras procedentes de cordón, se forman células adultas capaces de secretar concentraciones adecuadas de enzima α -L-iduronidasa para la metabolización del material de depósito acumulado, incluso con mayor rapidez que en los trasplantes de células adultas. Según un estudio realizado durante 7 años en 20 pacientes afectados de síndrome de Hurler 22 (media de edad de 16 meses) se produce una estabilización e incluso un incremento de la función neurocognitiva, alcanzando a los 72 meses índices similares a la normalidad. También mejoran la dismorfia facial, las visceromegalias, el desarrollo psicomotor y se detiene la progresión de la miocardiopatía y la hepatopatía característica. Sin embargo, la efectividad del tratamiento en las alteraciones musculoesqueléticas requiere mayor número de estudios para poder confirmarse. La supervivencia es del 85 % con persistencia de actividad enzimática normal en sangre periférica a los 7 años (media de recuperación medular 24-56 días).
En los TMO se ha descrito un índice de fracaso del 28-37 % con la consiguiente disminución de la actividad enzimática en sangre periférica a pesar del uso previo de irradiación corporal total. En el caso del trasplante de células hematopoyéticas procedentes de cordón, la histocompatibilidad requerida es de menor grado que en el caso de TMO, lo que facilita la búsqueda de donante compatible (media: 15 días) y disminuye la mortalidad atribuible a la enfermedad de injerto contra huésped, que alcanza el 30-55 % de casos con un 50 % de mortalidad asociada en los TMO de donante no emparentado 23.
Debido a los efectos beneficiosos que aporta la temprana exposición a la α -L-iduronidasa, se ha propuesto el uso de la terapia enzimática sustitutiva previa a la realización del trasplante y después de él, hasta alcanzar la recuperación medular con actividad enzimática normal en sangre periférica. Se ha descrito una disminución de la sintomatología pretrasplante con reducción de la morbimortalidad asociada postrasplante. Sin embargo, ante la dificultad de predecir la respuesta que las IgG anti-α -L-iduronidasa originadas durante el tratamiento enzimático sustitutivo pudieran tener en la eficacia del trasplante posterior, su uso previo generalizado en el síndrome de Hurler debería continuar analizándose mediante ensayos clínicos controlados 24.
La aplicación de la transferencia génica, mediante retrovirus en el tratamiento de diversas enfermedades genéticas y metabólicas, se ha desarrollado enormemente en los últimos 15 años. Las células madre hematopoyéticas se han considerado excelentes receptores de la transferencia génica por su enorme capacidad de renovación y diferenciación en múltiples líneas celulares. Las limitaciones de la eficiencia de esta transferencia están siendo salvadas por la creación de nuevos vectores (virus adenoasociados, lentivirus) y avances en el procesamiento celular que otorga a las células hematopoyéticas mayor capacidad de transducción 25.
Según estudios recientes realizados en tejido cerebral de ratones, la inyección del gen que codifica la α -L-iduronidasa mediante virus adenoasociados que actúan como vectores, ha resultado beneficiosa con disminución del material de depósito de la distensión lisosomal y su desaparición en el tejido cerebral en la 26 semana postinyección. Así mismo, puede transferirse mediante retrovirus la propia α -L-iduronidasa, expresando posteriormente las células del receptor concentraciones enzimáticas correctas con la consiguiente metabolización y normalización del depósito de GAG en tejidos periféricos. Se ha observado que las células madre hematopoyéticas modificadas genéticamente son capaces de atravesar y corregir los defectos enzimáticos de fibroblastos no modificados, mediante transferencia proteica. Los resultados obtenidos sugieren la posibilidad de considerar la terapia génica como tratamiento futuro capaz de prevenir la enfermedad asociada al síndrome de Hurler y detener el deterioro neurocognitivo característico de estos pacientes 26,27.
En conclusión, señalamos que la terapia enzimática sustitutiva es eficaz y segura en el tratamiento de la MPS-I (síndrome de Hurler), con las limitaciones comentadas anteriormente. En aquellos casos en los que exista un donante compatible, está indicado realizar un trasplante de médula ósea o de células madre hematopoyéticas posterior al tratamiento enzimático. La terapia génica será probablemente el tratamiento del futuro.
