

Los tumores del SNC son los tumores sólidos más frecuentes en la edad pediátrica. Dentro de ellos los gliomas de bajo grado constituyen el tipo más común de tumor del SNC en niños, representando hasta el 30-50% de los mismos.
Pacientes y métodosAnálisis retrospectivo de las características epidemiológicas, manifestaciones clínicas, localización del tumor, histología, tipo de tratamiento si lo ha recibido, evolución y secuelas a largo plazo de 111 pacientes diagnosticados de glioma de bajo grado en el Hospital Infantil Universitario Niño Jesús de Madrid entre enero de 2002 y diciembre de 2011.
ResultadosDe los 111 pacientes 57 eran niños y 54 niñas. La edad media fue de 7,26 años (intervalo 2 meses-19 años). Los síntomas de presentación más frecuentes fueron la cefalea (27%) y los vómitos (19%). Las localizaciones más frecuentes fueron los hemisferios cerebrales (38%), seguido del tronco cerebral (27,4%) y del cerebelo (18,5%). Se realizó estudio histológico en 89 pacientes (80,18%), siendo el astrocitoma pilocítico el tipo histológico más frecuente. Se realizó biopsia diagnóstica en 20 pacientes (22,5%), resección parcial en 38 pacientes (42,7%) y resección total en 31 pacientes (34,8%). Recibieron quimioterapia 16 pacientes (14%) y radioterapia 18 pacientes (16%). La supervivencia global fue del 88,3%. Un paciente presentó secuelas auditivas, 5 pacientes presentaron secuelas visuales y 4 pacientes secuelas endocrinas.
ConclusionesEl tipo histológico más frecuente es el astrocitoma pilocítico. La supervivencia global fue del 88,3%. Solo el 9% de los pacientes presentaron algún tipo de secuela auditiva, visual o endocrinológica.
Central nervous system (CNS) tumors are the most common solid tumors in children. Among these, the low-grade gliomas are the most common type, accounting for up to 30-50% of them.
Patients and methodsA retrospective analysis was carried out on the epidemiology, clinical characteristics, tumor location, histology, treatment, outcome and long-term sequelae of 111 patients diagnosed with low-grade glioma in the Niño Jesús Children's Hospital of Madrid from January 2002 to December 2011.
ResultsOf the 111 patients, there were 57 boys and 54 girls. The mean age was 7.26 years (range, 2 months - 19 years). The most common symptoms of presentation were headache (27%) and vomiting (19%). The most common locations were the cerebral hemispheres (38%), followed by the brainstem (27.4%), and cerebellum (18.5%). Histological examination was performed in 89 patients (80.18%). Pilocytic astrocytoma was the most common histological type. Diagnostic biopsy was performed in 20 patients (22.5%), partial resection in 38 patients (42.7%), and total resection in 31 patients (34.8%). Sixteen patients received chemotherapy (14%), and eighteen patients received radiotherapy (16%). Overall survival was 88.3%. Long term hearing, visual and endocrine sequelae were note in 1, 5, and 4 patients, respectively.
ConclusionsThe most common histological type is pilocytic astrocytoma. Overall survival was 88.3%. Only 9% of patients had some kind or auditory, visual or endocrine sequelae.
Los tumores del sistema nervioso central (SNC) son los tumores sólidos más frecuentes en la edad pediátrica. Dentro de ellos los gliomas son, sin duda, el subtipo más común representando según las series del 46 al 70% de todos ellos1.
Los gliomas son un grupo heterogéneo de tumores que se clasifican histológicamente según los criterios de la Organización Mundial de la Salud (OMS) publicados en el año 2007 en 2 grupos, los de bajo y los de alto grado2. En la población pediátrica los gliomas de bajo grado son los más frecuentes, representando hasta un 30-50% de todos los tumores del SNC3–6.
Según la clasificación de la OMS los gliomas de bajo grado se pueden, a su vez, clasificar como tumores de grado 1 (astrocitoma pilocítico) o de grado 2 (astrocitoma fibrilar) (tabla 1)2. Aunque el cerebelo es el lugar de presentación más frecuente, pueden aparecer en cualquier lugar del SNC. Los síntomas que producen dependen de la localización y suelen estar presentes meses antes del diagnóstico. El tratamiento de elección es la cirugía y la resección completa es el factor pronóstico más importante para la supervivencia.
Clasificación de los gliomas de bajo grado según la OMS
| Tumores astrocitarios |
| Grado 1 |
| Astrocitoma pilocítico |
| Astrocitoma subependimario de células gigantes |
| Grado 2 |
| Astrocitoma difuso (fibrilar, gemistocítico o protoplásmico) |
| Astrocitoma pilomixoide |
| Xantoastrocitoma pleomórfico |
| Tumores oligodendrogliales |
| Grado 2 |
| Oligodendroglial |
| Tumores neuronales y neurogliales mixtos |
| Grado 1 |
| Ganglioglioma |
| Gangliocitoma |
| Ganglioglioma desmoplásico infantil |
| Tumor neuroepitelial disembrioplásico |
En esta revisión hemos realizado un análisis retrospectivo de 111 pacientes pediátricos con gliomas de bajo grado diagnosticados en el Hospital Infantil Universitario Niño Jesús durante un periodo de 10 años.
Pacientes y métodosSe realizó un estudio retrospectivo de los pacientes diagnosticados de glioma de bajo grado en el Servicio de Oncología Pediátrica del Hospital Infantil Universitario Niño Jesús de Madrid durante un periodo de 10 años, entre el 1 de enero de 2002 y el 31 de diciembre de 2011.
En cada uno de los pacientes se recogieron los datos epidemiológicos (edad, sexo), manifestaciones clínicas, tiempo transcurrido hasta el diagnóstico, localización del tumor, histología, tipo de tratamiento si lo ha recibido, evolución y secuelas a largo plazo. Se incluyeron pacientes que cumpliesen los siguientes criterios de inclusión: a) edad al diagnóstico ≤ 20 años; yb) tumor glial de grado i o ii según la clasificación de la OMS o glioma de bajo grado según los hallazgos radiológicos en la RMN. Se diagnosticaron de gliomas de bajo grado aquellos tumores que en la RMN se presentaban como lesiones intraaxiales, homogéneas, hipointensas en T1, hiperintensas en T2, sin o con mínima captación tras la administración de contraste. Además, en algunos casos se realizó RMN con difusión y/o espectroscopia, etiquetándose de gliomas de bajo grado aquellos que no mostraban restricción a la difusión y que no presentaban alteraciones en los picos de NAA, colina y lípidos, respectivamente7–9.
ResultadosEn el periodo de 10 años de estudio un total de 111 pacientes fueron diagnosticados de glioma de bajo grado en el Hospital Infantil Universitario Niño Jesús de Madrid. De los 111 pacientes 57 eran niños (51,4%) y 54 niñas (48,6%). La edad media al diagnóstico fue de 7,26 años, con una desviación estándar de 7 años (intervalo 0,16-18,66 años).
Los síntomas de presentación fueron en orden descendente de frecuencia: cefalea (27%), vómitos (19%), crisis comiciales (18%), ataxia (16%) y afectación de pares craneales (14%). Tres pacientes (2,7%) comenzaron con tortícolis y uno (0,9%) con alucinaciones visuales y auditivas. Cinco de los pacientes presentaban edema de papila en el momento del diagnóstico. En todos los casos había transcurrido más de un mes desde el inicio de los síntomas hasta el diagnóstico, y en el 37% habían transcurrido más de 6 meses. De los 111 pacientes 5 de ellos estaban diagnosticados de neurofibromatosis tipo 1 y 2 de esclerosis tuberosa.
En cuanto a la localización del tumor la más frecuente fue el cerebro (incluyendo vía óptica y región hipotalámica) con 43 pacientes (38%), seguido del tronco cerebral con 31 pacientes (27,4%), el cerebelo con 21 pacientes (18,5%), los ventrículos con 7 pacientes (6,2%) y la médula espinal con 7 pacientes (6,2%). En el momento del diagnóstico solo un paciente presentó diseminación leptomeníngea. Se trataba de un lactante de 2 meses de edad con astrocitoma hipotalámico confirmado histológicamente.
El diagnóstico se hizo por estudio histológico en 89 pacientes (80,18%), y en el resto, 22 pacientes (19,82%), por los hallazgos radiológicos en la RMN (previamente descritos). Los diagnósticos histológicos fueron 39 astrocitomas pilocíticos (43,8%), 14 gangliogliomas (15,7%), 5 tumores neurogliales (5,6%), 2 astrocitomas difusos (2,2%), 2 astrocitomas pilomixoides (2,2%), un xantoastrocitoma pleomórfico (1,1%), un astrocitoma subependimario de células gigantes (1,1%) y, por último, 25 tumores etiquetados como astrocitarios de bajo grado (28%).
De los 22 pacientes en los cuales no pudo realizarse estudio histológico, y cuyo diagnóstico se estableció en función de los hallazgos radiológicos (previamente descritos), 16 pacientes presentaban tumor al nivel del tronco cerebral, 3 pacientes tumor al nivel de nervio óptico, 2 pacientes tumor al nivel de la médula espinal y un paciente tumor en el ventrículo. Los 3 pacientes con glioma de nervio óptico, uno de ellos con afectación bilateral, estaban diagnosticados de neurofibromatosis tipo 1 y el paciente con tumor a nivel ventricular estaba diagnosticado de esclerosis tuberosa, siendo en este último caso la sospecha de un astrocitoma subependimario de células gigantes.
En lo relacionado con el tratamiento fueron sometidos a cirugía 89 pacientes. De ellos en 20 casos (22,5%) solo pudo realizarse biopsia con fines diagnósticos. De los 69 restantes en 38 pacientes (42,7%) se realizó resección parcial y en 31 pacientes (34,8%) fue posible la resección total. Recibieron quimioterapia 16 pacientes (14,4%). De ellos en 7 pacientes se había realizado resección parcial, en 4 solo biopsia diagnóstica y en 5 ningún tipo de cirugía. En cuanto al régimen quimioterápico empleado 10 pacientes recibieron quimioterapia según el protocolo SIOP vigente en ese momento, un paciente con glioma medular recibió cisplatino e irinotecán y en los restantes 5 pacientes se desconoce qué régimen se utilizó. Dieciocho pacientes (16,2%) recibieron radioterapia, siendo su edad media de 6,82 años (intervalo 3,25-10,08 años). Siete de estos pacientes presentaban tumor de tronco cerebral (biopsiados 4 de ellos) y recibieron radioterapia siendo menores de 8 años como segunda línea de tratamiento tras la quimioterapia. Los restantes 11 pacientes superaban esta edad en el momento de recibir la radioterapia como primera línea de tratamiento. De los 18 pacientes que recibieron radioterapia en 6 se había realizado resección parcial y en 5 biopsia diagnóstica. Un paciente diagnosticado de astrocitoma pilocítico quiasmático a los 10 meses recibió inicialmente quimioterapia; posteriormente a los 3 años presentó progresión tumoral y recibió radioterapia. Los pacientes que recibieron quimioterapia y/o radioterapia presentaban sintomatología derivada de la localización tumoral en el tronco cerebral, en la médula espinal o en la vía óptica o próxima a ella (paresia o parálisis de pares craneales y/o hemiparesia o disminución de la agudeza visual). Ninguno de los pacientes sometidos a resección tumoral total recibió quimioterapia o radioterapia posterior.
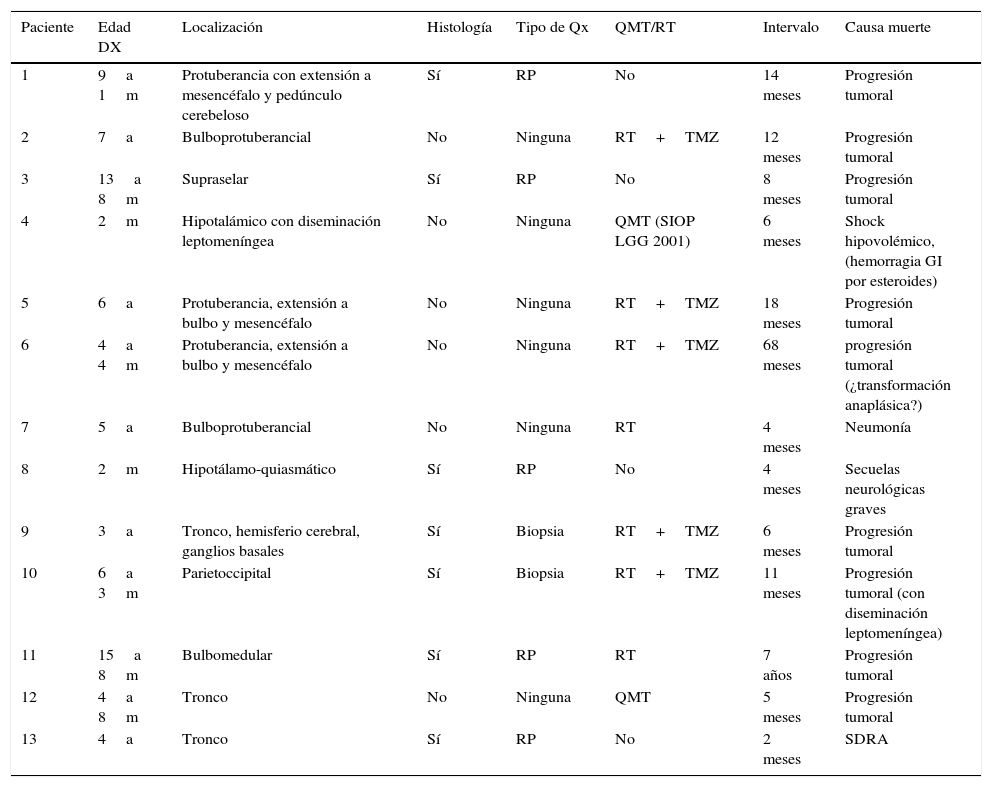
La supervivencia global fue del 88,3%, con una mediana de seguimiento de 4,08 años (intervalo: 0,04-9,72 años). En los 31 pacientes en los que se pudo realizar una resección completa del tumor la supervivencia fue del 100%, mientras que en los restantes 80 pacientes la supervivencia fue del 83,7%. La resección total se asoció con mayor supervivencia global (p<0,05). De los 13 pacientes fallecidos 5 habían sido sometidos a resección parcial (ninguno de ellos recibió quimio o radiopterapia), 2 solo biopsia (ambos recibieron radioterapia y temozolamida) y en los otros 6 no había sido posible ningún abordaje quirúrgico (uno recibió solo radioterapia, 2 recibieron quimioterapia y 3 radioterapia con temozolamida). La principal causa de muerte fue la progresión tumoral (tabla 2).
Características de los pacientes fallecidos
| Paciente | Edad DX | Localización | Histología | Tipo de Qx | QMT/RT | Intervalo | Causa muerte |
|---|---|---|---|---|---|---|---|
| 1 | 9a 1m | Protuberancia con extensión a mesencéfalo y pedúnculo cerebeloso | Sí | RP | No | 14 meses | Progresión tumoral |
| 2 | 7a | Bulboprotuberancial | No | Ninguna | RT+TMZ | 12 meses | Progresión tumoral |
| 3 | 13a 8m | Supraselar | Sí | RP | No | 8 meses | Progresión tumoral |
| 4 | 2m | Hipotalámico con diseminación leptomeníngea | No | Ninguna | QMT (SIOP LGG 2001) | 6 meses | Shock hipovolémico, (hemorragia GI por esteroides) |
| 5 | 6a | Protuberancia, extensión a bulbo y mesencéfalo | No | Ninguna | RT+TMZ | 18 meses | Progresión tumoral |
| 6 | 4a 4m | Protuberancia, extensión a bulbo y mesencéfalo | No | Ninguna | RT+TMZ | 68 meses | progresión tumoral (¿transformación anaplásica?) |
| 7 | 5a | Bulboprotuberancial | No | Ninguna | RT | 4 meses | Neumonía |
| 8 | 2m | Hipotálamo-quiasmático | Sí | RP | No | 4 meses | Secuelas neurológicas graves |
| 9 | 3a | Tronco, hemisferio cerebral, ganglios basales | Sí | Biopsia | RT+TMZ | 6 meses | Progresión tumoral |
| 10 | 6a 3m | Parietoccipital | Sí | Biopsia | RT+TMZ | 11 meses | Progresión tumoral (con diseminación leptomeníngea) |
| 11 | 15a 8m | Bulbomedular | Sí | RP | RT | 7 años | Progresión tumoral |
| 12 | 4a 8m | Tronco | No | Ninguna | QMT | 5 meses | Progresión tumoral |
| 13 | 4a | Tronco | Sí | RP | No | 2 meses | SDRA |
Once pacientes presentaron progresión tumoral y solo en uno de ellos se había realizado resección total. En uno de estos pacientes se sospechó transformación anaplásica. Se trata de una niña de 4 años con una tumoración al nivel del tronco cerebral que se extiende desde el mesencéfalo hasta el bulbo, diagnosticada según las imágenes de RMN de glioma de bajo grado, no abordable con cirugía. Recibió radioterapia con temozolamida concomitante, consiguiendo reducción del tamaño tumoral y mejoría clínica. Desde entonces el tumor permaneció estable hasta que, a los 9 años de edad, presenta datos de progresión clínico-radiológica, y dada la inusual evolución se decide realizar estudio histológico que confirma la presencia de glioblastoma multiforme. En este caso, dado que no se disponía de estudio histológico al diagnóstico, la transformación anaplásica fue una sospecha no confirmada.
Por último, en cuanto a las secuelas a largo plazo, un paciente con astrocitoma protuberancial (con estudio histológico) sometido a radioterapia presentó hipoacusia leve, 5 pacientes con glioma de la vía óptica presentaron disminución de la agudeza visual (4 de ellos habían recibido quimioterapia y uno radioterapia), 4 pacientes con gliomas supraselares o quiasmáticos desarrollaron alteraciones endocrinas (uno hipotiroidismo y diabetes insípida, uno panhipopituitarismo, otro pubertad precoz y el último panhipopituitarismo y diabetes insípida).
DiscusiónLos gliomas de bajo grado suelen tener un comportamiento indolente, incluso se han documentado casos de regresión espontánea en niños10,11, en contraste con lo que ocurre en pacientes adultos en los que suelen tener un comportamiento más agresivo. Su característico comportamiento indolente hace que casi el 50% de los pacientes tengan manifestaciones clínicas de 6 meses de evolución o más cuando son diagnosticados12. En nuestra serie había transcurrido más de un mes desde el inicio de los síntomas hasta el diagnóstico en todos los casos, y 6 meses o más en casi el 40% de los pacientes.
Las manifestaciones clínicas están determinadas por la localización e histología del tumor, la edad del niño y su desarrollo neurológico. La mayoría, tales como cefalea (típicamente matutina), náuseas, vómitos, disminución del nivel de conciencia, papiledema y parálisis del vi par se deben al aumento de la presión intracraneal por obstrucción de la circulación del líquido cefalorraquídeo. Por otra parte, hay síntomas/signos derivados de la localización del tumor. Así, los gliomas de cerebelo pueden provocar ataxia y dismetría, los gliomas hemisféricos convulsiones, hemiparesia o alteraciones del comportamiento, los gliomas hipotalámicos y de la glándula pituitaria obesidad, diabetes insípida, disfunciones endocrinas o alteraciones visuales, los gliomas de la vía óptica alteraciones visuales, proptosis o estrabismo, los gliomas de tronco cerebral alteraciones de pares craneales (por ejemplo disfagia, disartria) y afectación de vías largas (hemiparesia, espasticidad, hiperreflexia, signo de Babinski) y, por último, los gliomas cervicomedulares tortícolis, déficit sensitivo o afectación de vías largas3. En raras ocasiones pueden metastatizar o transformarse en gliomas de alto grado3,13,14. La diseminación o mestástasis se ha descrito en el 3-5% de los pacientes al diagnóstico y en el 7-10% de los pacientes durante el curso evolutivo del tumor, siendo más frecuente en niños menores de 1 año14–19. En la literatura la mayoría son casos aislados y alguna pequeña serie, la más grande de 13 pacientes14,16–21. En nuestra serie solo un paciente presentó diseminación leptomeníngea en el momento del diagnóstico, y solo en un paciente hubo sospecha de transformación anaplásica. El primer caso se trataba de un niño de 2 meses de edad con glioma parietoccipital biopsiado. El segundo caso presentaba un tumor de tronco, con extensión desde el mesencéfalo hasta el bulbo, que fue diagnosticado de glioma de bajo grado según las imágenes de RMN, sin estudio histológico. Se trató con radioterapia y temozolamida concomitante consiguiendo la disminución del tamaño tumoral y la mejoría clínica. El paciente permaneció estable hasta que, 5 años después del diagnóstico, presenta progresión clínico-radiológica y la biopsia realizada en ese momento confirma la presencia de glioblastoma multiforme. Algunos autores sugirieron una asociación entre la radioterapia y la transformación maligna22. Por otra parte, según un reciente estudio, los gliomas protuberanciales (sobre todo los difusos) son en su mayoría (hasta el 91%) gliomas de alto grado23. En nuestra paciente, al no disponer de estudio histológico en el momento del diagnóstico, la transformación anaplásica es una sospecha. El largo intervalo de tiempo entre el diagnóstico y la progresión tumoral, así como el antecedente de haber recibido radioterapia, apoyan que sí haya presentado transformación anaplásica, mientras que la localización tumoral en el tronco cerebral con afectación protuberancial va más a favor de que se tratase de un glioma de alto grado ya desde el inicio.
Históricamente el cerebelo es la localización más prevalente, de modo que los gliomas de bajo grado del cerebelo suponen del 15 al 25% de todos los tumores del SNC en niños. Están seguidos por los gliomas de los hemisferios cerebrales (10-15%), gliomas de estructuras de la línea media (10-15%), gliomas de la vía óptica (5%) y gliomas del tronco cerebral (2-4%)4. En nuestra casuística los hemisferios cerebrales (incluyendo el nervio óptico) y el tronco cerebral aparecen como las localizaciones más frecuentes, a diferencia de lo que se encuentra en la literatura.
Los niños con neurofibromatosis tipo 1 acumulan la mayoría (más del 70%) de los gliomas de la vía óptica e hipotalámicos24. De hecho, hasta el 15-20% de los niños con neurofibromatosis tipo 1 desarrollan gliomas de la vía óptica, aunque solo la mitad de ellos se harán sintomáticos y precisarán tratamiento25. En nuestra serie de los 5 pacientes diagnosticados de NF tipo 1, 3 presentaban gliomas de bajo grado del nervio óptico y uno de ellos bilateral.
Las técnicas de imagen (TC y preferiblemente RMN) permiten una aproximación diagnóstica inicial y será la biopsia la que confirme la histología tumoral. La RMN craneoespinal permite estudiar la presencia de diseminación leptomenínegea, que como ya se ha comentado es infrecuente, aunque si se sospecha deberá realizarse junto con el estudio citológico de LCR26. La realización de biopsia no siempre es posible, sobre todo en tumores localizados en el nervio óptico, en el tronco cerebral o en estructuras profundas. En este sentido, para preservar la función del nervio óptico los niños con gliomas de la vía óptica o de la región hipotalámica con imagen compatible con glioma de bajo grado, y sobre todo si están diagnosticados de neurofibromatosis tipo 1, no son sometidos a biopsia. En el caso de los gliomas de bajo grado de tronco cerebral o estructuras profundas la biopsia también se realiza con cautela, especialmente si son asintomáticos o si no presentan progresión en estudios de imagen seriados3. En nuestro caso solo se realizó biopsia en 7 de los 31 pacientes con sospecha de glioma de bajo grado de tronco, el resto fueron diagnosticados en función de los criterios radiológicos (previamente descritos). De los 5 pacientes con neurofibromatosis tipo 1 los 3 que presentaban tumor de la vía óptica sugestivo de glioma de bajo grado no fueron sometidos a biopsia; a los otros 2 pacientes, uno con tumor de hemisferio frontal y otro con tumor medular, sí se les realizó biopsia.
La base del tratamiento es la cirugía. Ha de realizarse resección completa del tumor siempre que sea posible, teniendo en cuenta las secuelas que puedan resultar de la misma. La quimioterapia y/o radioterapia se reserva para aquellos casos en los que la resección completa no haya sido posible y el paciente presente síntomas. Dado que son tumores de bajo grado, si el paciente permanece asintomático habrá que valorar con cautela estos tratamientos, ya que no están exentos de efectos secundarios27–30. La radioterapia se asocia con defectos neuroendocrinos y cognitivos, vasculopatía y segundos tumores, sobre todo en pacientes con neurofibromatosis tipo 127–29. Con el objetivo de evitar estas complicaciones, en los niños más pequeños se suele optar por la quimioterapia con menos efectos secundarios o de menor repercusión (por ejemplo alergia a carboplatino)30. En nuestra serie la mayoría de los pacientes que recibieron quimioterapia y/o radioterapia presentaban sintomatología derivada de la localización tumoral en el tronco cerebral, en la vía óptica o próxima a ella o en la médula espinal (paresia o parálisis de pares craneales y/o hemiparesia o disminución de la agudeza visual) y fueron los pacientes de mayor edad los que recibieron radioterapia como primera línea de tratamiento. Está ampliamente descrito que el grado de resección quirúrgica es el factor asociado más fuertemente con la supervivencia global y libre de progresión12,31–35, alcanzándose una supervivencia a los 10 años del 90% o mayor en el caso de la resección total12,26,31,33,36,37. Ninguno de nuestros pacientes en los que pudo realizarse resección total del tumor falleció.
La histología tumoral parece ser un factor de progresión independiente. Así los tumores no pilocíticos, especialmente el fibrilar difuso, se asocian con mayor tendencia a progresión, recurrencia y transformación anaplásica, aunque como se ha dicho esta última es rara en la edad pediátrica12,31,34,35,38. En nuestra serie 11 pacientes presentaron progresión tumoral, y solo en uno de ellos se sospechó transformación anaplásica, como ya se ha comentado.
Los niños tienen mejor supervivencia que los adultos, aunque el papel de la edad como factor pronóstico es incierto1,26,35,38–40.
En conclusión, la supervivencia de este tipo de tumores con los tratamientos actuales es alta, en nuestra serie global del 88,3% y en los que se realizó resección total del 100%. Por otra parte, la supervivencia con secuelas no debe considerarse necesariamente un éxito y, por ello, debemos minimizar las secuelas derivadas del tratamiento manteniendo siempre el objetivo riesgo/beneficio para intentar conseguir una buena calidad de vida. En nuestra serie, a pesar de un seguimiento corto, solo el 9% de los pacientes presentó algún tipo de secuela auditiva, visual o endocrinológica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
