

La enzima P450c17 cataliza 2 reacciones diferentes: 17α-hidroxilación de la progesterona y pregnenolona y segmentación de la unión del carbono 17-20 a partir de la 17,20liasa para producir andrógenos suprarrenales. Esta enzima está codificada por el gen CYP17A1. Se presenta una paciente de 14años con retraso en el desarrollo puberal y presión arterial elevada para su talla y edad. Cariotipo 46,XX. En el estudio hormonal destaca hipogonadismo hipergonadotropo, así como una insuficiencia suprarrenal y exceso mineralocorticoideo. El estudio genético mostró una mutación en homozigosis en el gen CYP17A1 (c.753+1G>A), no descrita previamente, la cual es responsable de la fisiopatología de la deficiencia de 17α-hidroxilasa. Esta entidad es una forma rara de hiperplasia suprarrenal congénita. Normalmente la enfermedad suele pasar desapercibida hasta la adolescencia o el inicio de la vida adulta y se debería sospechar ante individuos 46,XY con genitales ambiguos o 46,XX con retraso puberal que asocia hipertensión y/o hipopotasemia.
P450c17 enzyme catalyses two different reactions: the 17α-hydroxylation of progesterone and pregnenolone, and segmenting the carbon 17-20 binding from the 17,20lyase producing adrenal androgens. This enzyme is coded by the CYP17A1 gene. The case is presented of a 14 year old patient with delayed pubertal development and a high blood pressure for height and age. 46,XX karyotype. Hormonal studies highlighted hypergonadotropic hypogonadism, adrenal insufficiency and mineralocorticoid excess. Subsequent genetic studies showed a homozygous mutation in the CYP17A1 gene (c.753+G>A), not previously described, which is responsible for the pathophysiology of 17α-hydroxylase deficiency. This entity is a rare form of congenital adrenal hyperplasia. The disease often goes unnoticed until adolescence or early adult life, and should be suspected in 46,XY individuals with ambiguous genitalia or 46,XX with delayed puberty associated with hypertension and/or hypokalaemia.
La deficiencia de 17α-hidroxilasa (17-OHD) es una forma rara de hiperplasia suprarrenal congénita (HSC) con una herencia autosómica recesiva y una incidencia aproximada de 1:50.000-100.000 en recién nacidos1.
La enzima citocromo P450c17 está codificada por el gen CYP17, situado en el cromosoma 10q24.32. Contiene 8exones y se expresa en la corteza adrenal y las gónadas, pero no en la placenta ni en las células de la granulosa.
Las características clínicas típicas de la 17-OHD fueron descritas por primera vez en los años sesenta en una paciente con hipertensión arterial (HTA) crónica y pubertad retrasada en la que se demostraba hipopotasemia y actividad de renina y aldosterona suprimidas3. En la actualidad y desde la clonación del gen CYP17 se han descrito aproximadamente 50mutaciones diferentes4–6.
Se presenta una paciente con características clínicas y analíticas sugestivas de 17-OHD en la que se ha demostrado una mutación en el gen CYP17 todavía no descrita.
Caso clínicoPaciente mujer de 14años con retraso en el desarrollo puberal. No presenta antecedentes familiares ni personales de interés. En la exploración física se detectan talla de 160,3cm (–0,13SDS) y peso de 49,3kg (–0,57SDS), normales, destacando ausencia de signos puberales (T1, P1, A1) con genitales externos femeninos infantiles. La edad ósea se encuentra llamativamente retrasada (9años y 6meses) y la presión arterial (PA) es 130/75mmHg (p90-99 para su talla y edad), sin variaciones posturales ni asimetrías. Se realiza un estudio hormonal inicial en el que aparecen gonadotropinas muy elevadas (LH31,92 y FSH121,49mUI/ml) con estradiol disminuido (<10pg/ml), así como un exceso de ACTH (228pg/ml) y progesterona (11,40ng/ml) con niveles bajos de cortisol (0,80μg/dl), situándonos por lo tanto en el contexto de un hipogonadismo hipergonadotropo acompañado de insuficiencia suprarrenal. En el resto de los parámetros bioquímicos destaca únicamente un potasio en el límite bajo de la normalidad (3,8mg/dl) con otros electrolitos normales. Se realiza un cariotipo, resultando 46,XX, y el estudio del gen SRY es negativo. La ecografía ginecológica demuestra un útero rudimentario, sin poder identificarse ovarios.
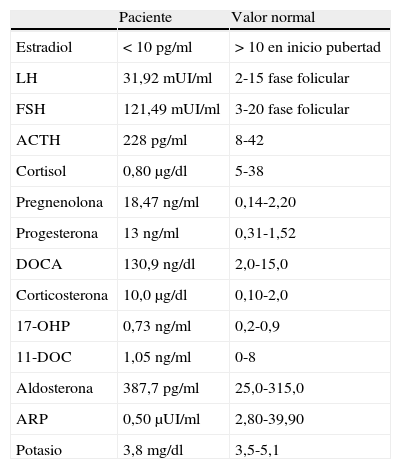
Ante los resultados obtenidos se amplía el estudio suprarrenal valorando el ritmo circadiano y se observa una pérdida del mismo con valores mantenidos de ACTH elevada y cortisol bajo en distintos momentos del día (ACTH262 y 585pg/ml y cortisol 0,49 y 0,10μg/dl a las 8:00 y las 16:00h, respectivamente). A continuación se analizan los distintos metabolitos intermedios de la vía del cortisol y se encuentra una elevación de pregnenolona, progesterona, desoxicorticosterona (DOCA), corticosterona y aldosterona, así como una actividad de renina plasmática suprimida. Los resultados y sus valores de referencia se presentan en la tabla 1.
Estudio hormonal de la paciente y los valores de normalidad
| Paciente | Valor normal | |
| Estradiol | <10 pg/ml | > 10 en inicio pubertad |
| LH | 31,92 mUI/ml | 2-15 fase folicular |
| FSH | 121,49 mUI/ml | 3-20 fase folicular |
| ACTH | 228 pg/ml | 8-42 |
| Cortisol | 0,80μg/dl | 5-38 |
| Pregnenolona | 18,47 ng/ml | 0,14-2,20 |
| Progesterona | 13 ng/ml | 0,31-1,52 |
| DOCA | 130,9 ng/dl | 2,0-15,0 |
| Corticosterona | 10,0μg/dl | 0,10-2,0 |
| 17-OHP | 0,73 ng/ml | 0,2-0,9 |
| 11-DOC | 1,05 ng/ml | 0-8 |
| Aldosterona | 387,7 pg/ml | 25,0-315,0 |
| ARP | 0,50μUI/ml | 2,80-39,90 |
| Potasio | 3,8 mg/dl | 3,5-5,1 |
ACTH: hormona adrenocorticotropa; ARP: actividad de renina plasmática; DOCA: desoxicorticosterona; FSH: hormona foliculoestimulante; LH: hormona luteinizante; 17-OHP: 17- hidroxiprogesterona; 11-DOC: desoxicortisol.
Finalmente se realiza estudio genético, encontrándose una mutación en homocigosis (c.753+1G>A) en el lugar dador del splicing del exón 4 del gen CYP17A1, que no ha sido descrita en la actualidad.
En el momento del diagnóstico de presunción se inicia tratamiento con hidrocortisona vía oral 10mg/m2/día y parches de estrógenos. Los parámetros analíticos de insuficiencia suprarrenal se han ido corrigiendo progresivamente y se normalizan las cifras de potasio y la PA. Inicia botón mamario a los 5meses del tratamiento y mantiene un buen desarrollo puberal. A los 17años, y tras alcanzar el desarrollo mamario completo, se añade al tratamiento gestágenos, iniciando menstruaciones regulares. La paciente presentaba llamativa osteoporosis, que ha mejorado con el tratamiento sustitutivo, pasando la Z-score de DMO en columna lumbar de –4,3 a –2,8SDS. En la actualidad tiene 18años, talla de 168,8cm (0,74SDS), peso de 65kg (0,65SDS), no presenta clínica sugestiva de insuficiencia suprarrenal y no han aparecido otras incidencias en la evolución.
DiscusiónLa síntesis de estrógenos forma parte de la esteroidogénesis que se produce en la corteza suprarrenal y que viene definida por unas reacciones enzimáticas que permiten la conversión del colesterol en diversos esteroides biológicamente activos que pertenecen a uno de los siguientes grupos principales: mineralocorticoides, glucocorticoides y andrógenos7. Como es previsible por su origen embriológico común, las células esteroidogénicas de la corteza suprarrenal y las gónadas comparten una porción significativa de la maquinaria celular responsable de la biosíntesis de esteroides y conforman en su conjunto la ruta esteroidogénica (fig. 1).

La paciente presenta un defecto en la 17α-hidroxilasa, con las características clínicas y hormonales típicas. Normalmente la enfermedad suele pasar desapercibida hasta la adolescencia o el comienzo de la vida adulta. La combinación de hipogonadismo e insuficiencia suprarrenal con el exceso mineralocorticoideo nos hacen sospechar el diagnóstico de 17-OHD. La presentación clásica es HTA, hipopotasemia y pubertad retrasada con falta de desarrollo de los caracteres sexuales secundarios en las mujeres3. En los varones pueden aparecer diferentes grados de genitales ambiguos con falta de virilización. Aproximadamente el 90% presentan HTA o hipopotasemia en el momento del diagnóstico.
El gen CYP17 codifica la enzima P450c17, que se encarga de catalizar 2 reacciones enzimáticas: la 17α-hidroxilación de la progesterona y pregnenolona, y la segmentación de la unión del carbono 17-20 a partir de la 17,20liasa para producir dehidroepiandrosterona y androstenediona en la glándula suprarrenal y gónadas. Alteraciones en CYP17 resultan en un deterioro de la producción de cortisol con hipersecreción secundaria de ACTH y producción de grandes cantidades de DOCA y corticosterona, que causan un estado de exceso de mineralocorticoide caracterizado por HTA, hipopotasemia y alcalosis. En consecuencia, el eje renina-angiotensina queda suprimido, provocando un defecto de síntesis de aldosterona que conlleva un hipoaldosteronismo hiporreninémico. Pese al deterioro de la producción de glucocorticoides, las concentraciones elevadas de corticosterona protegerían una posible insuficiencia suprarrenal y no aparecen síntomas clásicos de la enfermedad de Addison que ocurren en la mayoría de defectos enzimáticos suprarrenales8. El tratamiento con glucocorticoides en estos pacientes corrige la HTA y la hipopotasemia, de forma que suprime el exceso mineralocorticoideo inducido por el exceso de ACTH. Por otro lado, el defecto en la producción de andrógenos suprarrenales se traduce en una falta de estrógenos y explica el hipogonadismo hipergonadotropo, de manera que aparece ausencia de vello púbico y axilar, así como falta de desarrollo mamario y amenorrea. La terapia con estrógenos inicialmente induce la pubertad en estas pacientes y se añade progesterona más adelante para regular los ciclos menstruales, previniendo de este modo también una osteoporosis en la vida adulta9.
El diagnóstico hormonal de 17-OHD se basa en la elevación de DOCA, corticosterona y pregnenolona, metabolitos no disponibles en todos los laboratorios. Sin embargo, la medición de progesterona suele ser más sencilla, y en un estudio de pacientes con mutaciones en CYP17, todos ellos presentaban niveles basales elevados de 0,7 a 14ng/ml comparados con personas normales8; por tanto, podría ser un marcador sensible de la enfermedad.
La confirmación diagnóstica se realiza por estudio molecular del gen CYP17. Desde las primeras mutaciones identificadas por Kagimoto et al.4, se han descrito más de 59mutaciones en el gen CYP17, incluyendo simples cambios de bases, inserciones y deleciones. La localización más frecuente en la región del exón8. La mayoría representan mutaciones esporádicas (www.hgmd.cf.ac.uk), de manera que 17-OHD supone aproximadamente el 1% de los casos de HSC del mundo, pero en algunos grupos étnicos aparece una prevalencia mucho más elevada y se ha sugerido un efecto fundador, como la microinserción de 4 nucleótidos en el codón480 descritos en los descendientes menonitas canadienses10, la deleción de la fenilalanina en el codón53 o 54 descrito en japoneses11, una microdeleción de 9 nucleótidos cubriendo los codones487-489 en asiáticos12 y varias mutaciones en población brasileña5,8.
En conclusión, se ha presentado una paciente mujer con una mutación en homocigosis del gen CYP17 que no ha sido descrita todavía, la cual es responsable de la fisiopatología de 17-OHD. Esta enfermedad rara deberíamos tenerla en cuenta ante individuos 46,XY con genitales ambiguos o 46,XX con retraso puberal en los que se asocia HTA y/o hipopotasemia. La identificación de nuevas mutaciones en CYP17 es importante a la hora de comprender los mecanismos moleculares de su deficiencia y aportar información sobre la estructura enzimática de P450c17.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Presentado como póster en el Congreso SEEP 2011, Granada.
