



Introducción
La hipobetalipoproteinemia se define por la presencia de niveles séricos de colesterol total (CT) por debajo del percentil 5 para la edad y sexo, aproximadamente 150 mg/dl, lipoproteínas de baja densidad (c-LDL), aproximadamente 70 mg/dl, y de apoproteína B (apo-B), aproximadamente 50 mg/dl1. Estas alteraciones analíticas pueden ser secundarias a dietas vegetarianas o a diversas enfermedades como la malabsorción intestinal, patologías hepáticas, malnutrición o hipertiroidismo, entre otras. Dentro de las causas primarias, además de la hipobetalipoproteinemia familiar (HBF), debemos descartar la abetalipoproteinemia causada por mutaciones en el gen de la proteína microsomal de transferencia de triglicéridos (MTP), que se encarga de transferir lípidos a la apo-B (4q22-q24), dotándola de la conformación adecuada para la formación de lipoproteínas2.
La HBF es una alteración que se hereda con un patrón heterogéneo, en la que los portadores heterozigotos son asintomáticos y presentan concentraciones circulantes de apo-B y c-LDL por debajo de la normalidad, por lo que pueden estar protegidos, en cierto modo, frente a la aterosclerosis y sus complicaciones. Los portadores homozigotos3, por el contrario, tienen gran variedad de manifestaciones clínicas4,5 derivadas de la absorción intestinal deficiente de grasa y de vitaminas liposolubles.
Una minoría de casos de HBF, de herencia autosómica dominante, son secundarios a mutaciones en el gen APOB1,2,6 (2p24), que dan origen a la traducción de una apo-B anómala que altera la síntesis de las lipoproteínas que la contienen. En estos casos, para llegar al diagnóstico definitivo es necesario realizar la secuenciación del gen APOB7,8.
Se presenta el caso de una familia con cuatro miembros afectados de HBF debido a una mutación en heterozigosis en el gen APOB.
Observación clínica
Varón de 8 años y 7 meses de edad remitido para estudio por el hallazgo casual de concentraciones plasmáticas de CT, c-LDL y triglicéridos (TG) disminuidas.
Entre los antecedentes familiares destacaban la presencia en madre, abuela y tía materna de hipocolesterolemia, con 2 hermanos sanos y con cifras normales de CT y TG. El resto de antecedentes familiares carecían de interés.
En la exploración física presentaba buen estado general, hábito asténico, buena coloración de piel y mucosas, peso de 24,3 kg (P25) y talla de 132,6 cm (P75). Abdomen blando, depresible, sin masas ni visceromegalias palpables, soplo sistólico de características funcionales. Genitales externos masculinos normales, testes en bolsas de 2 ml de Prader; estadio puberal I de Tanner. Resto de la exploración normal.
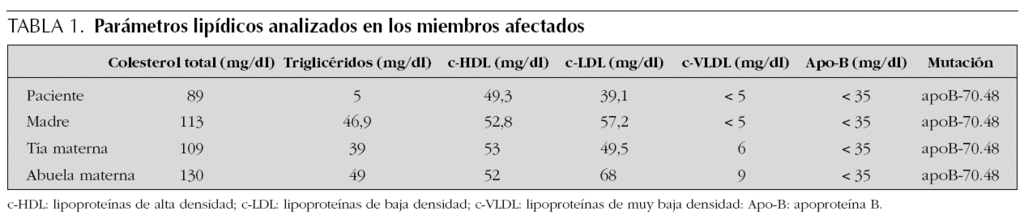
Los análisis realizados mostraron un CT de 89 mg/dl (v.n. 120-220 mg/dl), TG: 3 mg/dl (v.n. 25-115 mg/dl), c-LDL: 39,1 mg/dl (v.n. 60-130 mg/dl), lipoproteínas de muy baja densidad (c-VLDL) < 5 mg/dl (v.n. 5-23 mg/dl) y apo-B < 35 mg/dl (v.n. 30-100 mg/dl), con lipoproteínas de alta densidad (c-HDL) de 49,3 mg/dl (v.n. 35-65 mg/dl). Los niveles de vitamina E fueron < 4,9 mg/l (v.n. 4,9-2 mg/l). El resto de los valores analíticos, incluyendo vitamina A, se encontraron dentro de límites normales.
En los estudios realizados a la madre, abuela y tía materna, se observó hipocolesterolemia con cifras de TG normales (tabla 1). En el resto de miembros de la familia, incluidos padre y hermanos del paciente, no se encontraron alteraciones del lipidograma.
La secuenciación del gen APOB del paciente y sus familiares descubrió la existencia tanto en él como en la madre, abuela y tía materna, de una mutación (G → T) en el exón 26. Esta sustitución convierte el codón GAA (que codifica ácido glutámico) en posición 3198 en un "stop codon" (TAA). Esta mutación sin sentido conduce a la formación de una apo-B truncada de 3.197 aminoácidos que determina la formación de una proteína correspondiente al 70,48 % de la Apo B 100 madura (4.536 aminoácidos). Todos ellos presentaban la mutación en heterozigosis y se encontraban asintomáticos.
El paciente permanece asintomático y sólo recibe tratamiento con suplementos de vitaminas E.
Discusión
Nuestro caso presenta disminución de los niveles de CT, TG, c-LDL, c-VLDL y apo-B, así como deficiencia de vitamina E. Así mismo, los familiares afectados muestran hipocolesterolemia y descenso de apo-B con cifras normales de TG y vitamina E, sin síntomas asociados.
Los pacientes heterozigotos, en general, no precisan un tratamiento específico9, salvo suplementos de vitamina A y E en caso de deficiencia, como en nuestro caso índice, para prevenir las posibles alteraciones neurológicas10,11.
En los portadores homozigotos las manifestaciones clínicas12,13 son heterogéneas y de diversa severidad, pudiendo dar origen a un cuadro clínico similar al de la abetalipoproteinemia congénita o enfermedad de Bassen-Kornzweig. Puede existir malabsorción de grasas y déficit de vitaminas liposolubles (principalmente A y E) por acumulación de TG en el enterocito, que no se absorben adecuadamente al no formarse los quilomicrones. Todo ello produce acantocitosis, síntomas neurológicos (vértigos, migraña y ataxia, entre otros) y degeneración retiniana, debidas al déficit de vitamina E, cuyo transporte en plasma requiere apo-B. En estos casos es fundamental la instauración de un tratamiento precoz con aporte de vitaminas A y E y reducción de la ingesta de grasa.
Debido a todas las posibles complicaciones descritas asociadas a las formas homozigotas, consideramos de gran importancia el estudio genético familiar en los pacientes y sus familiares14,15. En el caso que describimos se descartó la existencia de formas homozigotas, y dicho estudio permitió realizar consejo genético a los miembros afectados de esta familia.
Hay descritas múltiples alteraciones moleculares que dan origen a HBF. Esta entidad se ha visto asociada a mutaciones tanto en el cromosoma 2 como en el 3, existiendo familias en las que no se detectan alteraciones en ninguno de estos 2 cromosomas14. Hay también diferentes mutaciones descritas en el gen de la Apo-B, en nuestro caso se trata de la denominada apoB-70.48 (que da origen a una proteína correspondiente al 70,48 % de la Apo B 100 madura)8. La denominada apoB-38.7, por otra parte, se asocia a un cuadro de hipobetalipoproteinemia severo, diabetes mellitus tipo 2 y calcificaciones arteriales.
Debido a la infrecuencia de esta enfermedad y a su probable infradiagnóstico en los sujetos asintomáticos, el interés de nuestro caso se centra en la realización de estudio genético de esta familia, demostrándose la existencia de una misma mutación en todos los miembros afectados de forma heterozigota.
Agradecimientos
Correspondencia: Dra. M.ªT. Muñoz Calvo.
Servicio de Endocrinología. Hospital Infantil Universitario Niño Jesús.
Avda. Menéndez Pelayo, 65. 28009 Madrid. España.
Correo electrónico: munozmaite@yahoo.es
Recibido en noviembre de 2006.
Aceptado para su publicación en enero de 2007.
Al Prof. S Calandra, de la Universidad de Módena & Reggio Emilia (Italia) por la realización del estudio molecular de los pacientes.
