

La hipofosfatasia (HF), descrita en 1948 por Rathbun1, es una enfermedad congénita, caracterizada por un defecto en la mineralización ósea y dentaria, secundario a una deficiencia en la biosíntesis de la isoenzima tisular inespecífica de la fosfatasa alcalina ósea, hepática y renal (TNSALP)2,3.
Su incidencia en población europea se estima en 1/300.000 recién nacidos para las formas graves y en 1/6.370 en las moderadas4.
Su expresión clínica es muy variable, desde casos de muerte intraútero por alteración severa de la mineralización ósea, a casos con caída precoz de la dentición en la edad adulta exclusivamente. Dependiendo de la edad de aparición de los síntomas, se diferencian 6 formas clínicas, con severidad y pronósticos muy diferentes, y que pueden solaparse entre sí: perinatal letal, perinatal benigna, infantil, juvenil, adulta y odontohipofosfatasia2,5.
Se presenta el caso clínico de un varón, en estudio desde los 11 meses de edad por retraso del desarrollo psicomotor (ausencia de gateo y bipedestación estable). Carecía de antecedentes familiares de relevancia en relación con alteraciones óseas y dentición, sin consanguinidad. Su gestación se produjo mediante fecundación in vitro (implantación de embriones con semen paterno). Se detectó acortamiento femoral en ecografía prenatal a las 20 semanas de edad gestacional. Su cariotipo (amniocentesis) fue 46 XY. El parto, a término (semana 38+5), fue cesárea por oligoamnios, con un peso al nacimiento de 2.270g (-2,5 DE) y una longitud de 45cm (-2,6 DE para su edad gestacional). Recibió suplementos orales de vitamina D3 (400 UI/día) desde el nacimiento. Presentó un cierre precoz de la sutura coronal izquierda, en cuyo estudio se detectó una malformación de Arnold Chiari tipo 1. No había tenido fracturas óseas, pero sí un retraso de la dentición (presencia exclusiva de incisivos inferiores y superior derecho a los 15 meses).
A la edad de 17 meses, mostraba: longitud: 73,9cm (-2,65 DE), peso: 8,4kg (-2,43 DE) y perímetro cefálico: 49cm (+2 DE para edad-talla), con dolicocefalia, frente prominente, fontanela anterior de 2×3cm, raíz nasal aplanada, hipertelorismo, filtrum alargado y paladar ojival, tórax campaniforme con pectus excavatum y ensanchamiento del extremo distal de ambos antebrazos.
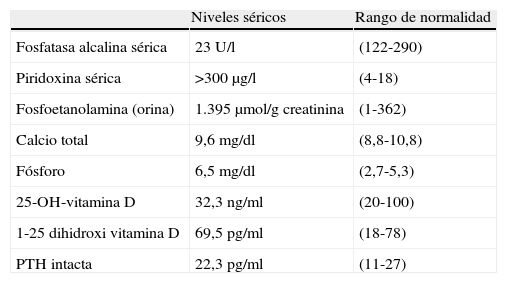
La serie ósea radiológica mostró malformaciones metafisarias de huesos largos (fig. 1) y ensanchamiento de costillas. El estudio bioquímico confirmó la existencia de niveles disminuidos de fosfatasa alcalina sérica, junto con un notable incremento de los niveles sus sustratos: piridoxina y fosfoetanolamina (formas inorgánicas de fósforo), con niveles normales de 25-OH-vitamina D y del resto de parámetros del metabolismo fosfocálcico estudiados (tabla 1). La secuenciación del gen codificante de la fosfatasa alcalina (ALPL), detectó la presencia en heterocigosis compuesta de las mutaciones conocidas c.542C>T (p.S181L) y c.644T>C (p.I215T), confirmándose el diagnóstico de hipofosfatasia.
, que se encuentran ensanchadas, con pérdida de densidad ósea, trabéculas groseras y proyecciones radiolucentes que se extienden desde la fisis hacia la metáfisis. Reacción perióstica lineal en el radio izquierdo.")
Marcada alteración en metáfisis de huesos largos (proximales de ambos húmeros, distales de ambos radios y cúbitos y proximales de ambos fémures, tibias y peronés), que se encuentran ensanchadas, con pérdida de densidad ósea, trabéculas groseras y proyecciones radiolucentes que se extienden desde la fisis hacia la metáfisis. Reacción perióstica lineal en el radio izquierdo.
Resultados del estudio del metabolismo fosfo-cálcico
| Niveles séricos | Rango de normalidad | |
| Fosfatasa alcalina sérica | 23 U/l | (122-290) |
| Piridoxina sérica | >300μg/l | (4-18) |
| Fosfoetanolamina (orina) | 1.395μmol/g creatinina | (1-362) |
| Calcio total | 9,6mg/dl | (8,8-10,8) |
| Fósforo | 6,5mg/dl | (2,7-5,3) |
| 25-OH-vitamina D | 32,3ng/ml | (20-100) |
| 1-25 dihidroxi vitamina D | 69,5pg/ml | (18-78) |
| PTH intacta | 22,3pg/ml | (11-27) |
Los pacientes con HF infantil pueden desarrollar sus manifestaciones durante los primeros 6 meses, tras haber nacido asintomáticos, como en este caso, con un pronóstico vital favorable. Las complicaciones respiratorias secundarias a malformaciones torácicas y el aumento de la presión intracraneal por craneosinostosis precoz, determinan una tasa de mortalidad del 50% en los primeros meses2,5,6.
Los estudios radiológicos ponen de manifiesto una disminución generalizada de la densidad ósea y alteraciones metafisarias en los huesos largos similares a las que aparecen en formas graves de raquitismo2,5,6. Diferencialmente con este último, los niveles de fosfatasa alcalina sérica están disminuidos en la HF, acompañados de una elevación de las concentraciones de sus sustratos fosforados no degradados: fosfoetanolamina (PEA), piridoxal-5′-fosfato (PLP) y pirofosfatos inorgánicos (PPi)2,5,6. En general, puede afirmarse que «cuanto más severa es la enfermedad, menores son los niveles de fosfatasa alcalina2,6». En los casos más graves, se puede hallar hiperfosfatemia e hipercalciuria con o sin hipercalcemia2.
La enfermedad es debida a mutaciones del gen ALPL, que codifica la isoenzima tisular no-específica de fosfatasa alcalina (TNSALP)2,7. El diagnóstico definitivo lo permitirá la secuenciación del gen (1p36.1-p34) formado por 12 exones8. Presenta gran heterogenicidad alélica y se han descrito más de 190 mutaciones, lo que explica los distintos grados en la actividad de la enzima, condicionando la gran variabilidad en la expresión clínica de la enfermedad2,9. La variabilidad en la herencia, con patrones autosómicos recesivos y dominantes, con penetrancia variable, hace complicado el consejo genético2.
Aún carecemos de tratamiento curativo para la hipofosfatasia, empleándose tratamientos sintomáticos y medidas ortopédicas para intentar minimizar las manifestaciones de la enfermedad10. No obstante, el desarrollo de un tratamiento enzimático sustitutivo, en fase de ensayo actualmente, ha mostrado resultados esperanzadores.
