






La incontinencia pigmenti es una genodermatosis poco frecuente, de herencia ligada al cromosomaX, que afecta a tejidos derivados del ectodermo. Nuestro objetivo es revisar de la forma más completa posible los casos diagnosticados en edad pediátrica en dos hospitales.
Material y métodosSe ha realizado un estudio transversal retrospectivo, recogiéndose datos clínicos, analíticos, radiológicos y genéticos valorados a nivel multidisciplinar de pacientes diagnosticados en la edad pediátrica de incontinencia pigmenti desde el año 2004 al 2018.
ResultadosSe incluyeron 13pacientes diagnosticados de incontinencia pigmenti, todas de sexo femenino. Se realizó estudio genético en 11 de las 13, confirmándose alteraciones compatibles en 10 de ellas. Se observó afectación extracutánea relacionada con la enfermedad a nivel neurológico (con alteraciones radiológicas en 6 casos y expresión clínica en 3 de ellas), oftalmológico (4 casos), odontológico (7 casos) y hematológico (4 casos).
ConclusionesPresentamos el estudio más completo publicado hasta ahora de incontinencia pigmenti en España. Los resultados del estudio de las manifestaciones de la enfermedad fueron similares a las series de casos más amplias publicadas a nivel internacional y refuerzan la importancia de un estudio y seguimiento multidisciplinar.
Incontinentia pigmenti is a rare genodermatosis of inheritance linked to the Xchromosome that affects tissues derived from ectoderm. The aim of the study is to review, as completely as possible, the cases diagnosed in paediatric patients in two hospitals.
Material and methodsA retrospective cross-sectional study was carried out, using the clinical, analytical, radiological, and genetic data of paediatric patients diagnosed with incontinentia pigmenti from 2004 to 2018. The data collected were analysed and evaluated at a multidisciplinary level.
ResultsA total of thirteen patients diagnosed with incontinentia pigmenti were included in the study. All of them were female. A genetic study was performed on 11 patients, which confirmed findings compatible with incontinentia pigmenti in 10 of them. Extracutaneous involvement associated with the disease was observed at neurological level (radiological findings in 6 cases, and clinical expression in 3 of them), ophthalmological level (4 cases), dental level (7 cases), and haematological level (4 cases).
ConclusionsA presentation is given of the most complete study published so far of incontinentia pigmenti in Spain. In this study, the results of the disease manifestations were similar to the largest case series published internationally, which reinforces the importance of a multidisciplinary study and follow-up.
La incontinencia pigmenti (IP) es una genodermatosis poco frecuente, de herencia ligada al cromosomaX con carácter dominante y una penetrancia del 100%. Produce una afectación de tejidos derivados del ectodermo, presentándose como anomalías a nivel de piel, pelos, dientes, ojos y sistema nervioso1.
El primer caso fue descrito por Garrod en el año 1906 y cuenta con una incidencia aproximada de 1:40.000-50.000 recién nacidos, aunque la prevalencia no se conoce con exactitud ya que sigue siendo una patología infradiagnosticada1,2.
El comienzo clínico de la enfermedad se expresa generalmente a nivel cutáneo y durante las 2 primeras semanas de vida, afectando en más del 95% de los casos al sexo femenino1. Entre estas manifestaciones cutáneas se distinguen 4 fases: vesicular (I), verrugosa o liquenoide (II), hiperpigmentada (III) e hipopigmentada (IV)3. El inicio y la duración de cada estadio es diferente en cada individuo, pudiendo solaparse en el tiempo e incluso no manifestarse algunas de las fases4.
El gen mutado responsable de la enfermedad es el IKBKG (previamente conocido como NEMO), localizado en el locus Xq28. Este gen codifica una proteína (IKK-gamma o NEMO) que activa el factor nuclear-kappa B (NF-kB), el cual se encarga de regular la respuesta inmune, la cascada inflamatoria y los mecanismos de apoptosis celular. Entre el 80-90% de los casos la mutación se produce por deleciones de los exones 4 a 10 en el gen IKBKG, el resto se debe a otras alteraciones (missence, nonsense, cambio de marco, microdeleciones…) que se descubren gracias a la secuenciación del mismo2,5.
Se trata de un síndrome letal para el sexo masculino, salvo en situaciones como la presencia de mosaicismo, síndrome de Klinefelter (47XXY) o mutaciones hipomorfas en el gen IKBKG6.
En el año 1993, Landy y Donnai propusieron por primera vez una serie de criterios clínicos diagnósticos de IP; más tarde, Minić et al. los actualizaron, incluyendo el estudio genético7,8 (tabla 1).
Criterios diagnósticos de Landy y Donnai actualizados por Minić et al.7
| Criterios mayores | Criterios menores | Condiciones para confirmar el diagnóstico |
|---|---|---|
| Cambios típicos en la piel que se distribuyen en las líneas de Blaschko:• (Etapa I) Vesículo-bullosas• (Etapa II) Verrugosas• (Etapa III) Hiperpigmentada• (Etapa IV) Atrófica/hipopigmentada | Anomalías en sistema nervioso central/neurológicas: convulsiones, parálisis espástica, retraso psicomotor, retraso mental, microcefalia, atrofia cerebral/cerebelar, microgiria/polimicrogiria, hipoplasia en cuerpo calloso, alteración en ganglios de la base, leucomalacia periventricular, hidrocefalia, porencefalia, accidente cerebrovascular isquémico, necrosis hemorrágica difusa, encefalomielitisAnomalías oculares: defectos en la visión, retinopatía, desprendimiento de retina, alteración vascular en retina, anormalidades pigmentarias hiper/hipopigmentado, atrofia óptica, hipoplasia foveal, fibroplasia retrolental, cataratas, microftalmia, estabismo,nistagmoAnomalías dentales: retardo en la erupción primaria, anodoncia/hipodoncia, microdoncia, distrofia dental, anomalías de la forma (cónicos), impactación, diastema, malaoclusiónAlteraciones en paladar: paladar altoAlteraciones de la glándula mamaria: pezón supernumerarioAnormalidades en pelo (pelo, cejas, pestañas): alopecia, hipertricosisAnormalidades en uñas: distrofia, pigmentación amarillenta, hendiduras transversales o longitudinalesAbortos de fetos de sexo masculinoHallazgos histopatológicos típicos en piel | No existe evidencia de IP en familiar femenino de primer gradoSi no hay disponibilidad de realizar estudio molecular, se requieren 2 o más criterios mayores o un criterio mayor y uno menor para confirmar el diagnósticoMutación en IKBKG con cualquier criterio mayor o menor confirma el diagnósticoEvidencia de IP en un familiar femenino de primer grado: necesita un criterio mayor o 2 criterios menoresEosinofilia e inactivación del cromosoma X sesgada apoyan el diagnóstico en todos los casos |
El desconocimiento de esta patología hace que muchos casos leves pasen inadvertidos y lleguen a la edad adulta sin diagnóstico, y con ello se favorece la transmisión de la enfermedad a la descendencia. Además, en ocasiones se realiza un diagnóstico, pero no el cribado y seguimiento de las posibles alteraciones asociadas a nivel cutáneo y extracutáneo. En España se han publicado otras series de casos de IP, pero con menor número de pacientes y sin realizar un estudio tan amplio y completo de la enfermedad4,6,9-11.
El objetivo principal de este trabajo fue revisar de la forma lo más completa posible las manifestaciones clínicas de los casos de IP diagnosticados en la infancia en 2 centros hospitalarios españoles y comparar dichos datos con lo publicado a nivel internacional. Dicho objetivo va enfocado a generar en el personal sanitario más capacidad de sospecha clínica para realizar un diagnóstico más precoz de la enfermedad y favorecer la coordinación de un plan de seguimiento multidisciplinar de estos pacientes.
Material y métodosSe incluyeron pacientes diagnosticados en la edad pediátrica de IP mediante estudio genético y/o criterios clínicos modificados por Minić et al.7, por el servicio de Dermatología del Hospital Costa del Sol (Marbella) y del Hospital Regional Universitario Materno Infantil (Málaga), desde el año 2004 al 2018. Se realizó un estudio transversal retrospectivo recogiéndose datos clínicos, analíticos, radiológicos y genéticos de cada paciente desde la sospecha clínica inicial hasta la última revisión (diciembre de 2018): se incluyeron datos referentes a edad de comienzo de las lesiones cutáneas típicas y su evolución, otras posibles alteraciones cutáneas, de pelos, dientes y uñas, hemograma, realización de estudio genético, hallazgos histológicos (biopsia de piel), pruebas de neuroimagen y afectación clínica neurológica.
La sospecha clínica de IP estuvo siempre guiada por las lesiones cutáneas, que fueron el primer signo, apareciendo en todas en la etapa neonatal. A partir de ello se realizó un estudio anatomopatológico y genético en la mayoría de los casos, estableciéndose el diagnóstico de IP siguiendo los criterios diagnósticos ya comentados7.
El estudio fue iniciado y coordinado desde los servicios de Dermatología y Neuropediatría a través de las consultas y del área de hospitalización, realizándose una valoración de las pacientes a nivel multidisciplinar (por parte de neonatología, dermatología, neuropediatría, radiología, oftalmología y genética). Los procedimientos utilizados en los pacientes y la recogida de estos datos fueron realizados tras la obtención del consentimiento informado de los padres.
ResultadosSe recogieron un total de 13 pacientes diagnosticados de IP, todas de sexo femenino. El seguimiento de la evolución clínica de cada paciente se realizó hasta diciembre de 2018, salvo en los casos números 3 y 6, en que se perdió el seguimiento.
Ocho de las pacientes fueron valoradas por el servicio de Dermatología en los 30 primeros días de vida y las 5 restantes antes del año de vida. La edad de presentación de las manifestaciones cutáneas en todos los casos fue en la época neonatal. Todas debutaron con lesiones en fase vesiculosa siguiendo la distribución de las líneas de Blaschko, presentando en 2 casos un solapamiento con lesiones en fase hiperpigmentada (caso 3) y verrugosa (caso 9).
La evolución de las lesiones cutáneas fue variada, alternándose y solapándose en el tiempo las diferentes fases, e incluso sin aparente manifestación de alguna de ellas (fig. 1).
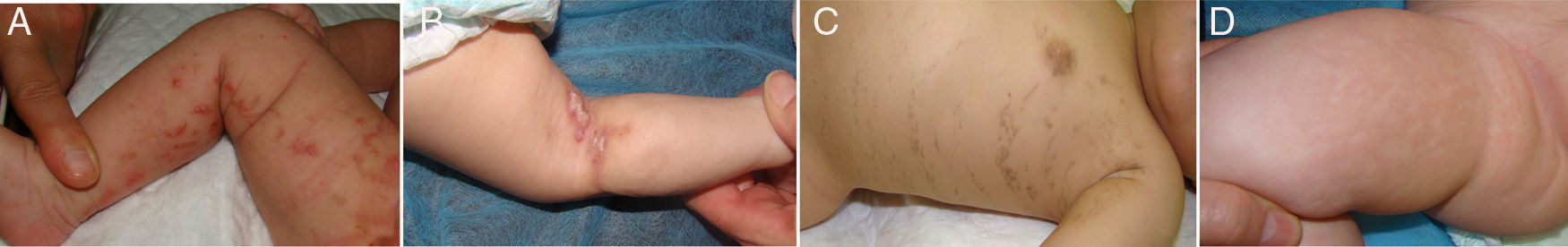
Se realizó tratamiento tópico de las lesiones cutáneas en las pacientes con mayor actividad inflamatoria. Se aplicaron corticoides en 4 casos (1, 2, 9 y 10) y de forma combinada con ácido fusídico en un caso (5), presentando buena respuesta a estos (tabla 2).
Manifestaciones dermatológicas de la serie de casos
| Casos | Edad de inicio lesiones cutáneas | Primera valoración por Dermatología | Inicio cutáneo | Evolución cutánea | Tratamiento de lesiones cutáneas | Otras alteraciones en piel/anejos | Estudio genético (edad de realización) | Biopsia piel (edad de realización) |
|---|---|---|---|---|---|---|---|---|
| 1 | Primeras 24 h | Periodo neonatal | Fase vesiculosa (vesículas en miembros inferiores [MMII], superiores [MMSS] y tronco que siguen líneas de Blaschko) | 1) Fase verrugosa2) Fase atrófica. | Corticoide tópico | Hemangioma infantil focal superficia. | 2 meses: deleción en exones 4-10 del gen IKBKG | 6 días: vesícula intraepidérmica eosinofílica |
| 2 | Primeras 24 h | Periodo neonatal | Fase vesiculosa (vesículas en MMII, menos evidentes que hermana de caso 1) | Fase hiperpigmentada (no fase verrugosa evidenciada) | Corticoide tópico | • Hemangioma infantil focal superficial• Mamilas supernumerarias | 7 meses: deleción en exones 4-10 del gen IKBKG | 6 días: muestra no valorable por escasa celularidad |
| 3 | 7 días | Periodo neonatal | Fase vesiculosa y fase hiperpigmentada (vesículas en tronco, MMII y cuero cabelludo e hipercrómicas con distribución blaschkoide en MMII) | Fase hiperpigmentada (no fase verrugosa evidenciada) | No | No realizado | 17 días: compatible con IP | |
| 4 | Primeras 24 h | 1 mes y 15 días | Fase vesiculosa (vesículas de distribución blaschkoide en MMII y raíz MMSS) | 1) Fase verrugosa.2) Fase hiperpigmentada[Reagudización ocasional en fase verrugosa] | Alopecia en vértex | 6 meses: deleción en exones 4-10 del gen IKBKG. De novo | 1 mes: espongiosis eosinofílica | |
| 5 | Primeras 24 h | Periodo neonatal | Fase vesiculosa (vesículas a nivel axilar y dorsal con distribución blashkoide) | Fase verrugosa e hiperpigmentada[Reagudización de lesiones en fase verrugosa con episodio de gastroenteritis aguda] | Ácido fusídico y corticoide tópico | • Pelo escaso y ralo en cuero cabelludo• Uñas finas y aplanadas | 19 meses: deleción en exones 4-10 del gen IKBKG | 1 mes: espongiosis con numerosos eosinófilos intravesiculares y dérmicos |
| 6 | Primeras 24 h | Periodo neonatal | Fase vesiculosa (lesiones versículo-pustulosas en pie izquierdo) | 1) Fase verrugosa.2) Fase hiperpigmentada | No | No realizado | No realizada | |
| 7 | 3 días | Periodo neonatal | Fase vesiculosa (lesiones eritematopustulosas de distribución blaschkoide en hemicuerpo derecho) | Fase verrugosa | No | 2 meses: no mutaciones en secuenciación completa del gen IKBKG en sangre (técnica PCR y MLPA) y muestra insuficiente de tejido cutáneo | 10 días: espongiosis con ampolla intraepidérmica con eosinófilos | |
| 8 | Primeras 24 h | 6 meses | Fase vesiculosa (vesículas de distribución blaschkoide en hemicuerpo izquierdo) | 1) Fase hiperpigmentada2) Desaparición progresiva de lesiones3) Fase atrófica | No | 11 meses: deleción en exones 4-10 del gen IKBKG | No realizada | |
| 9 | Primeras 24 h | Periodo neonatal | Fase vesiculosa y verrugosa (lesiones vesículo-costrosas de distribución lineal en piernas y brazos) | 1) Fase hiperpigmentada y verrugosa2) Fase hiperpigmentada y atrófica | Corticoide tópico | Hirsutismo en pubis secundario a esteroides tópicos prolongados | 7 meses: deleción en exones 4-10 del gen IKBKG. De novo. | 28 días: compatible con IP |
| 10 | Primeros 30 días | 3 meses | Fase vesiculosa (lesiones vesículo-ampollosas lineales siguiendo líneas de Blaschko) | 1) Fase verrugosa e hiperpigmentada2) Fase hiperpigmentada4) Fase verrugosa.5) Fase atrófica | Corticoide tópico | No | 3 meses: deleción en exones 4-10 del gen IKBKG | 7 días: aislados queratinocitos necróticos (resultado dudoso)14 días: compatible con IP |
| 11 | 15 días | 10 meses | Fase vesiculosa (vesículas en MMII de distribución blaschkoide) | 1) Fase verrugosa.2) Fase hiperpigmentada3) Desaparición progresiva de lesiones | No | 2 años: deleción en exones 4-10 del gen IKBKG | No realizada | |
| 12 | Primeras 24 h | 5 meses | Fase vesiculosa (vesículas de distribución lineal blaschkoide en MMII, perigenital y tórax derecho) | Fase hiperpigmentada | No | 5 meses: Variante p.GLn268Ter en el gen IKBKG | No realizada | |
| 13 | Primeros 30 días | Periodo neonatal | Fase vesiculosa (vesículas de distribución lineal blaschkoide en hemitórax derecho, MMII y dorsal) | Fase hiperpigmentada | No | 2 meses: Variante p.GLn268Ter en el gen IKBKG | No realizada |
Las alteraciones dermatológicas no cutáneas derivadas de la IP (pilosas y ungueales) se objetivaron solo en 3 casos (tabla 2).
Los estudios de neuroimagen mediante ecografía cerebral, tomografía computarizada (TC) y resonancia magnética (RM) craneal se realizaron en todos los casos, y se detectó afectación cerebral en 6 pacientes, presentando solo expresión clínica en 3 de ellas. En 2 de las pacientes no se apreciaron alteraciones en la ecografía cerebral inicial (casos 4 y 10), pero sí en RM cerebral realizada con posterioridad.
Presentaron afectación neurológica en forma de crisis convulsiva 3 pacientes y se observó alteración del desarrollo psicomotor en las mismas, y no en el resto de pacientes (tabla 3).
Manifestaciones a otros niveles de la serie de casos
| Casos | Pruebas de neuroimagen y función cerebral (edad de realización) | Afectación neurológica / Desarrollo psicomotor | Oftalmológico | Odontológica | Odontológica | Tiempo de seguimiento (edad) |
|---|---|---|---|---|---|---|
| 1 | • Ecografía craneal (4 días y 15 días): signos de vasculopatía calcificante en una arteria lenticuloestriada• Resonancia magnética (RM) craneal (1 mes): normal | No / Normal | Sin alteraciones | Dientes cónicosAgenesia dental | Eosionofilia | 3 años y 6 meses |
| 2 | Ecografía craneal: normal | No / Normal | Sin alteraciones | Dientes cónicos | Eosinofilia | 3 años y 6 meses |
| 3 | • Ecografía craneal (20 días): lesión hiperecogénica frontoparietal izquierda compatible con gliosis o isquemia• RM craneal (6 días): lesiones isquémicas subagudas en sustancia blanca de ambos hemisferios cerebrales en territorios limítrofes de la arteria cerebral anterior (ACA) y media (ACM) con mayor afectación izquierda• EEG (10 días): focalidad paroxística de punta onda en región frontal del hemisferio derecho | Periodo neonatal: Estatus convulsivo focal, secundario a ictus isquémico en territorio arteria cerebral anterior y media bilateral de mayor afectación izquierda / Hemiparesia izquierda | Sin alteraciones | No valorable (pérdida de seguimiento) | Eosinofilia | 1 mes y 17 días (pérdida de seguimiento) |
| 4 | • Ecografía craneal (4 meses): normal• RM craneal (3 años): lesiones subcentimétricas en cápsula interna de sustancia blanca de ambos hemisferios• RM craneal (5 y 7 años): lesiones en sustancia blanca a nivel subcortical y centro semioval izquierdo | No / Normal | Sin alteraciones | Retraso de erupción dentaria (1 año y 10 meses)Dientes cónicos | Normal | 9 años y 3 meses |
| 5 | • Ecografía craneal (2 y 17 días): dudosa hemorragia intraventricular (HIV) del plexo coroideo izquierdo con dilatación leve del ventrículo lateral izquierdo• Ecografía craneal (1 mes y medio): HIV resuelta, sin alteraciones• RM craneal (1 año y medio): normal | 3 meses de vida: Crisis convulsiva generalizada / Retraso global del desarrollo psicomotor | Retinopatía óptica del prematuro grado I en ojo derecho resuelta al año de vida | Normal | Normal | 4 años |
| 6 | Ecografía craneal (20 días): normal | No / Normal | Sin alteraciones | No valorable (por pérdida de seguimiento) | Eosinofilia | 20 días (pérdida de seguimiento) |
| 7 | Ecografía craneal (24 h y 30 días): normal | No / Normal | Sin alteraciones | Normal | Normal | 1 año y 3 meses |
| 8 | Ecografía craneal (7 meses): normal | No / Normal | Sin alteraciones | Normal | Normal | 10 años y 8 meses |
| 9 | RM craneal (1 año y 2 meses): normal | No / Normal | Lesión hemorrágica-isquémica en retina periférica nasal de ojo izquierdo | Normal | Normal | 12 años |
| 10 | • Ecografía craneal (7 días): normal• RM craneal (15 días): lesiones de baja intensidad serpenteantes en lóbulo parietal y occipital derechos en posible relación con malformación vascular de tipo venoso• EEG: focalidad paroxística parietotemporal derecha | Periodo neonatal: hemiconvulsión izquierda. Epilepsia focal sintomática / Hemiparesia izquierda | Desprendimiento total de retina ojo izquierdoMiopía magna en ojo derecho | Agenesia dental | Normal | 13 años y 6 meses |
| 11 | Tomografía computarizada (TC) craneal: normal | No / Normal | Sin alteraciones | Dientes cónicos | Normal | 14 años y 10 meses |
| 12 | RM craneal: afectación inespecífica difusa de sustancia blanca periventricular frontal y occipital bilateral | No / Normal | Tortuosidad vascular y manchas hipopigmentadas en ambas retinas | Hipodoncia | Normal | 2 años y 7 meses |
| 13 | • RM craneal: normal• EEG: normal | No / Normal | Ojo derecho: hemorragia retinianaOjo izquierdo: hiperpigmentación retiniana inespecífica | Dientes cónicosAgenesia dental | Normal | 2 años y 9 meses |
Durante el seguimiento por Oftalmología se apreció alteración en 5 casos, con afectación retiniana en todos ellos, relacionándose una de ellas con la prematuridad (tabla 3).
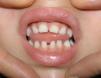
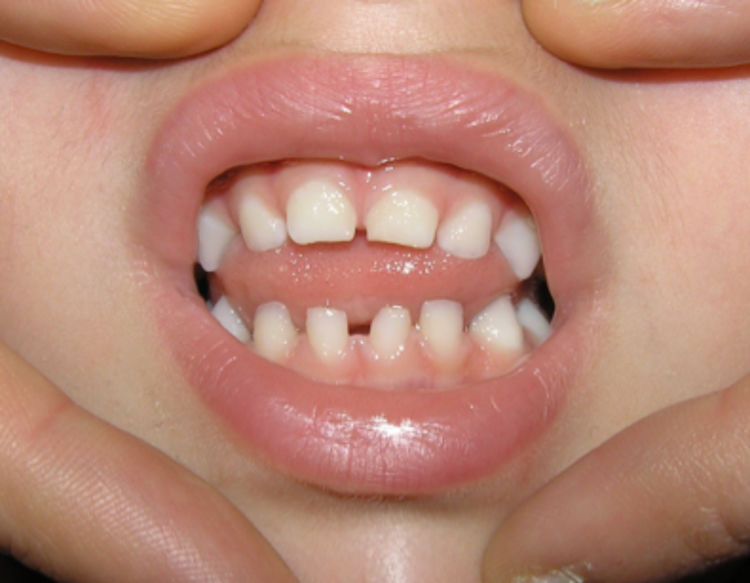
Se apreciaron alteraciones odontológicas en 7 de las 11 pacientes seguidas (en los casos 3 y 6 se perdió el seguimiento previamente al desarrollo de la dentición), identificándose alteración en número, retraso de inicio de dentición o de la morfología (tabla 3 y fig. 2).
Se encontraron alteraciones analíticas significativas (eosinofilia) sin otros hallazgos en 4 pacientes (casos 1, 2, 3 y 6) (tabla 3).
El estudio genético en muestra de sangre periférica se realizó en 11 pacientes: 8 presentaron deleciones de los exones 4 a 10 del gen IKBKG (realizado mediante amplificación por PCR para identificar deleción en exones 4 a 10), en uno (caso 7) no se observaron alteraciones en dicho gen y en 2 (casos 12 y 13) presentaron la variante patogénica p.GLn268Ter (c.802C>T) en heterocigosis en el gen IKBKG.
El primer estudio genético realizado en el caso 13 (búsqueda de deleciones de exones 4 a 10 mediante Long-Range PCR) no presentó alteraciones. Por este motivo se realizó la secuenciación del gen IKBKG (análisis mediante método Sanger de los exones codificantes y regiones de splicing del gen IKBKG, y comparación con la secuencia de referencia NM_003639.3) y se detectó la variante patogénica p.GLn268Ter (c.802C>T), la cual produce un codón stop prematuro, dando lugar a una proteína truncada y posiblemente no funcional responsable de la enfermedad.
Los casos 12 y 13 fueron fruto de embarazo por fecundación in vitro por ovodonación procedentes de la misma clínica de reproducción. Este hecho hizo sospechar que ambos casos podrían proceder de la misma donante, lo que justificó que el estudio genético del caso12 fuera dirigido desde el principio a descartar la variante patogénica p.GLn268Ter (c.802C>T).
En el caso 7, ante su aspecto clínico en forma de mosaico (lesiones localizadas en un único segmento corporal) y con estudio genético en sangre negativo (secuenciación del gen IKBKG mediante amplificación por PCR de los exones codificantes y regiones de splicing, comparando con la secuencia de referencia NM_003639.3, y estudio de posibles deleciones o inserciones mediante Multiplex Ligation-dependent Probe Amplification [MLPA]), se intentó realizar estudio genético sobre la biopsia de piel, sin que fuese técnicamente posible.
La edad media de realización del estudio genético fue a los 8meses. La biopsia cutánea se llevó a cabo en 8pacientes, en todos ellos durante los primeros 30días de vida, siendo compatible (espongiosis eosinofílica) en todas con IP (tabla 2).
El estudio genético fue realizado en las madres de 9pacientes, presentando la misma mutación que los casos índice en 7 de ellas. Las 2 madres restantes presentaban diagnóstico clínico de IP y refirieron que el estudio genético (realizado en un laboratorio externo extranjero) fue compatible con la enfermedad, pero los informes no fueron aportados.
DiscusiónNuestro estudio incluye una serie de casos de IP donde se recoge de forma completa la evolución clínica de las pacientes, aportando al trabajo documentación fotográfica, genética y pruebas complementarias. En él destaca la amplia variedad de manifestaciones clínicas recogidas relacionadas con IP en los distintos órganos y aparatos.
La mutación en el gen IKBKG por deleciones en los exones 4-10 se objetivó en la mayoría de los casos estudiados, concordando con lo descrito en la literatura (80-90%)1,2. En nuestra serie, la secuenciación del gen IKBKG permitió identificar una mutación no descrita hasta la fecha (variante patogénica p.GLn268Ter [c.802C>T]). Este hecho refuerza la importancia de realizar la secuenciación de dicho gen en todos los casos en los que el estudio de deleciones en los exones 4-10 sea negativo.
En este trabajo cabe destacar la presencia de un caso con mosaicismo somático (caso 7). En estos casos, si el estudio genético en sangre mediante secuenciación completa del gen IKBKG (incluido el estudio de posibles deleciones o inserciones mediante MLPA) es negativo, se recomienda realizar el estudio genético en muestra de tejido cutáneo afecto por la enfermedad12.
Al tratarse de una enfermedad mortal para el sexo masculino, se debe sospechar en madres con abortos de repetición de fetos varones, como aparece en los antecedentes maternos de algunos de nuestros casos6,8,13.
Ante el patrón de herencia autosómica dominante ligada al cromosomaX se recomienda realizar el estudio genético a la madre en caso de que no haya sido estudiada previamente. No obstante, hay que tener en cuenta que en el 65% de los casos la mutación es de novo2. Este porcentaje es algo menor en nuestra serie, y solo apareció en 2 de las pacientes estudiadas (18%, casos 4 y 9).
Al igual que en otros estudios, la fase vesiculosa fue la primera manifestación cutánea que apareció en todos los casos al debut durante el periodo neonatal, asociada o no a otros estadios2,12.
Los brotes de lesiones cutáneas suelen aparecer sin causa aparente, pero ocasionalmente pueden verse asociados a un proceso infeccioso intercurrente, como se observó en uno de nuestros casos.
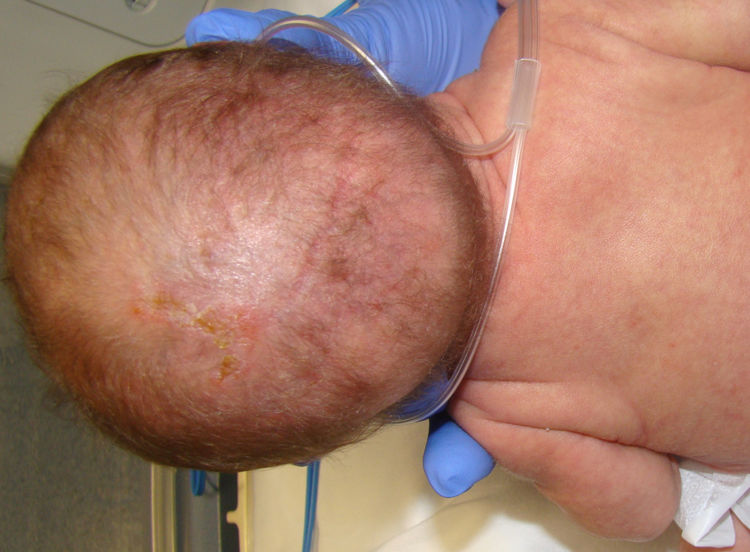
Las anomalías del pelo fueron evidenciadas solo en 2 de las pacientes (15,3%, casos 4 y 5) en forma de alopecia cicatricial y pelo ralo en cuero cabelludo, menor que en otras series (más del 50%)1-3,14 (fig. 3).
Las lesiones ungueales pueden afectar desde el 7-40% de los pacientes con IP, con una edad de aparición desde los 3 a los 45años (media de 26años)1,3,7. Solo se evidenció en una de nuestras pacientes (7%, caso 5); no obstante, son alteraciones que aparecen con la edad, por lo que aún habrá que continuar su seguimiento. A veces, en edades más tardías pueden aparecer tumores sub o periungueales queratósicos que producen dolor y deformidades por osteólisis de las falanges subyacentes y, en casos excepcionales; se ha asociado con la presencia de carcinomas escamosos subungueales1,3,7,12.
La afectación del sistema nervioso central (SNC) se manifiesta en el 30-50% de los pacientes con IP, apareciendo lesiones encefálicas heterogéneas (infartos, afectación de sustancia blanca y gris a varios niveles); aunque su patogénesis no está completamente aclarada, parece estar en relación con una vasculopatía cerebral derivada de la actividad inflamatoria que produce la enfermedad1,2,9,15. Estos datos se correlacionan con los resultados obtenidos en nuestro estudio, ya que el 50% de las pacientes que se realizaron RM cerebral (con técnica de difusión) presentaron alguna alteración.
La ecografía cerebral permite hacer una primera valoración en periodo neonatal; no obstante, la RM cerebral aporta mayor información, sobre todo si es con técnica de difusión2,15.
En ocasiones la ecografía cerebral inicial puede ser normal, pero en la RM cerebral posterior puede presentar lesiones, como en 2 de nuestros casos (4 y 10). Este hecho puede sugerir que los daños cerebrales aparecieran posteriormente en el tiempo entre la ecografía y la RM, falsos negativos de la técnica o interpretación de la ecografía, o puede deberse a que la RM sea una prueba más sensible que la ecografía. Por otro lado, destaca que en el caso 1 se observaron signos de vasculopatía calcificante mediante ecografía, pero la RM cerebral posterior fue normal. Estos datos defienden que la eco-TF no es suficiente para el estudio neurológico de los pacientes con IP, debiendo realizarse una RM cerebral lo más precoz posible. Algunos estudios recomiendan la realización de angio-RM para valorar con mayor exactitud la extensión de la vasculopatía15,16.
Las manifestaciones neurológicas se presentan en forma de crisis convulsivas, retraso mental, déficit motor (paresia/parálisis espástica) o microcefalia1,2,9,10,15. En el periodo neonatal la presentación clínica suele ser en forma de convulsiones o alteración del nivel de conciencia15.
En nuestra serie, solo 3 pacientes presentaron convulsiones (casos 3, 5 y 10), siendo estas mismas las únicas que desarrollaron algún otro tipo de alteración neurológica (2 con hemiparesia y una con retraso psicomotor).
Solo hubo correlación entre prueba de imagen y manifestación neurológica en 2 casos (3 y 10); el resto de las niñas con alteración en pruebas de imagen fueron asintomáticas a nivel clínico; no obstante, se trata de pacientes de corta edad, siendo necesario continuar el seguimiento de neurodesarrollo para esclarecer este hecho.
Las manifestaciones oculares aparecen en aproximadamente el 20-35% de los pacientes con IP (rango amplio según algunos estudios: 16-77%)1,7, asociándose frecuentemente a alteraciones en el SNC, con presencia de ceguera en el 7%. Se cree que el mecanismo que produce daño retiniano está en relación con fenómenos de isquemia resultantes de eventos vasooclusivos1,2,17.
En nuestro estudio se ha observado alguna afectación oftalmológica relacionada con IP en 4 casos (30%), encontrándose solo alteración neurológica asociada en uno de ellos (caso 10). Las pacientes que presentaron afectación retiniana requirieron en su mayoría tratamiento con láser y, una de ellas, cirugía por desprendimiento de retina unilateral, que finalmente precisó enucleación ocular. Hay que tener especial cuidado a la hora de diagnosticar pacientes con retinopatía del prematuro, como en el caso5, ya que suele confundirse con anomalías del desarrollo de vasos de la retina14. Las alteraciones no retinianas asociadas a IP (estrabismo, atrofia del nervio óptico, ptosis, hipoplasia del iris, pigmentación conjuntival, microftalmia, queratitis vorticial, cataratas, nistagmo y uveítis)1,14 no estuvieron presentes en estas pacientes.
Las alteraciones dentales aparecen en un 50-80%, en su mayoría en forma de dientes cónicos, seguido de hipodoncia, agenesia dental, retraso de la dentición y pérdida de piezas dentales1,11. Este hecho se correlaciona con los datos obtenidos en nuestro estudio (63% de las pacientes en las que fue posible la valoración odontológica). Existen otras anormalidades de la cavidad oral asociadas a IP, siendo las más frecuentes el paladar hendido y el paladar de arco alto, pudiendo asociar también maloclusión dental y asimetría facial1,2,11 (fig. 2).
Se han descrito alteraciones mamarias en el 11-30% de los pacientes con IP, siendo lo más frecuente la presencia de mamilas supernumerarias, además de hipoplasia o aplasia mamaria. En nuestra serie este hallazgo solo se presentó en una paciente en forma de mamila supernumeraria (caso 2)3,7,12.
Otro hallazgo asociado a IP es la presencia de leucocitosis con eosinofilia, sobre todo durante las fases cutáneas I y II7,12,17. En la revisión retrospectiva de nuestra serie se observó eosinofilia en 4 casos, de los cuales todos fueron durante el periodo neonatal, coincidiendo con las fases I y II en su mayoría. Dicho control analítico se realizó cuando los pacientes lo precisaban por procesos distintos a IP (riesgo infeccioso neonatal, procesos infecciosos…).
En diferentes estudios se han descrito casos de patologías de origen autoinmune, inmunodeficiencias y procesos neoplásicos malignos en pacientes con IP; este hecho se basa en el papel modulador del NF-kB3,18,19.
En nuestra serie no se detectaron dichas alteraciones asociadas; no obstante, es preciso realizar un seguimiento en el tiempo y vigilar su aparición.
Este estudio se ve limitado por la pérdida de seguimiento de varias de las pacientes, ya que se trasladaron a otras ciudades o no volvieron a acudir a consulta para su seguimiento; de la misma forma, la corta edad de las pacientes no permitió detectar alteraciones que aparecen con el paso de los años.
Se ha observado que el diagnóstico de IP es más precoz en los casos de los últimos 5años, probablemente gracias a un mayor conocimiento de la enfermedad. Es importante estructurar un plan seguimiento completo que se debe mantener a lo largo de la vida del paciente, incluido el consejo genético para futura descendencia16. La finalidad de este plan es detectar los signos y las patologías asociadas a IP, para así poder plantear un tratamiento dirigido lo más precoz posible y evitar complicaciones, sobre todo las alteraciones del SNC y oftalmológicas, que son las que determinan el pronóstico de la enfermedad2,10,20.
En conclusión, presentamos el estudio más completo publicado hasta ahora de IP en España, cuyos hallazgos fueron similares a las series de casos más amplias publicadas a nivel internacional, reforzando la importancia de conocer esta enfermedad y de realizar un estudio y un seguimiento multidisciplinar.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Departamento de Genética del Hospital Materno Infantil, Málaga, España.
