







Introducción
La piel constituye la barrera entre el medio interno y el medio externo. Durante el desarrollo del feto, la piel cambia el color, la consistencia, la función de barrera, las estructuras pilosebáceas y la cantidad de grasa subcutánea. Es por ello que el examen de la piel es una parte esencial en la estimación de la edad gestacional 1. La inspección de la piel, además de permitir establecer el diagnóstico de trastornos de la piel como órgano aislado e informar de la homeostasia corporal, puede detectar lesiones cutáneas que señalan la posibilidad de alteraciones del desarrollo del SNC y en muchas ocasiones ayuda a establecer el diagnóstico de entidades nosológicas específicas, principalmente síndromes genéticos. La epidermis y el cerebro derivan de la misma capa germinal, el ectodermo primordial, el cual se diferenciará en ectodermo de superficie y neuroectodermo (fig. 1). El ectodermo de superficie dará lugar a la epidermis y los apéndices cutáneos (pelo y uñas) y al esmalte de los dientes, mientras que del neuroectodermo se origina el SNC y sus apéndices, como la vesícula óptica, la neurohipófisis, la glándula pineal, así como la cresta neural. Esta última provoca células migratorias, progenitores multipotenciales, que contribuyen a la formación de diversos tipos de células, tejidos y elementos durante la embriogénesis 2. Este origen común del SNC y otros derivados ectodérmicos explica el porqué determinadas alteraciones cutáneas constituyen importantes indicios para el diagnóstico de entidades con riesgo neuroevolutivo.
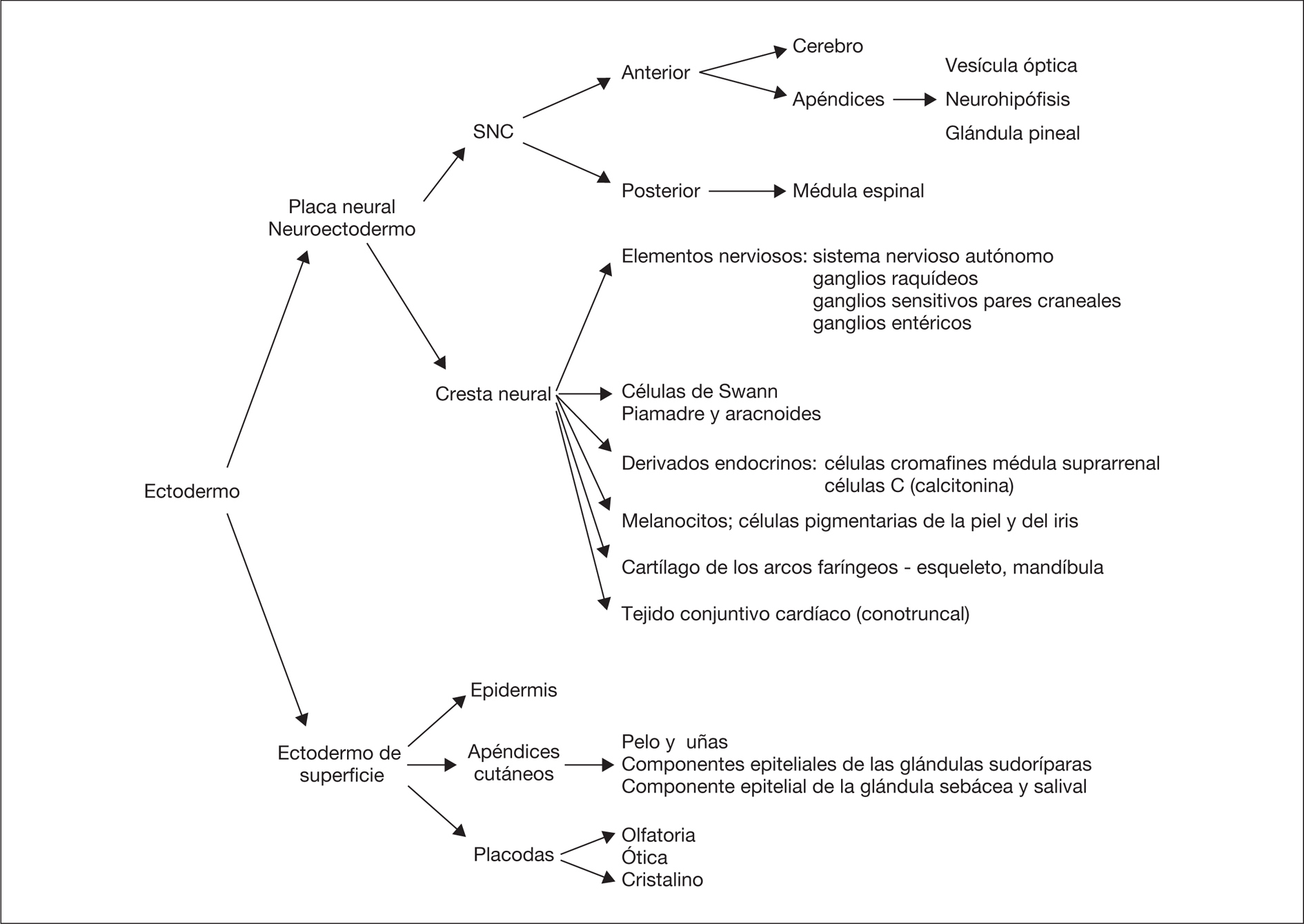
Figura 1. Principales estructuras derivadas del ectodermo.
Este particular enfoque no ha sido suficientemente tratado en la literatura médica neonatal, a pesar de que el conocimiento de estas alteraciones cutáneas ayuda a identificar rápidamente una población de recién nacidos con riesgo biológico de epilepsia y de trastorno motor y/o cognitivo. Esta identificación permite incluir a estos neonatos en programas de seguimiento de alto riesgo y formular planes de tratamiento y/o intervenciones terapéuticas. En algunos casos el diagnóstico posibilita el consejo genético a la familia. En este período de la vida se debe tener en cuenta que algunas alteraciones cutáneas de enfermedades congénitas pueden no estar presentes inmediatamente al nacer, pero se harán manifiestas durante las primeras semanas y meses de vida. Además, la expresión cutánea en algunas enfermedades varía con la edad, por lo que la sintomatología difiere de la observada en niños y adultos. Finalmente, la alteración del SNC puede expresarse en el período neonatal o bien posteriormente, durante la infancia o la niñez.
El objetivo de este artículo es facilitar el reconocimiento en el recién nacido de signos cutáneos que constituyen importantes pistas para el diagnóstico de entidades con riesgo de trastorno del neurodesarrollo.
Distribución de las lesiones cutáneas
Además de las características de la alteración cutánea, la distribución de esta puede aportar una importante información adicional necesaria para establecer la sospecha diagnóstica.
Distribución indicativa de mosaicismo cutáneo
Ciertas distribuciones señalan la existencia de un mosaicismo cutáneo (fig. 2). Estos cursan con un fenotipo heterogéneo que asocia con frecuencia anomalías extracutáneas, entre estas las neurológicas. Los mosaicismos cutáneos tienen generalmente presentación esporádica y pueden ser debidos a mosaicismos epigenéticos (inactivación X) y a mosaicismos genómicos que resultan de una mutación autosómica letal que sobrevive por mosaicismo 3. Según Happle las alteraciones de la piel en los mosaicismos cutáneos se manifiestan siguiendo cinco patrones de distribución 3 (fig. 2).
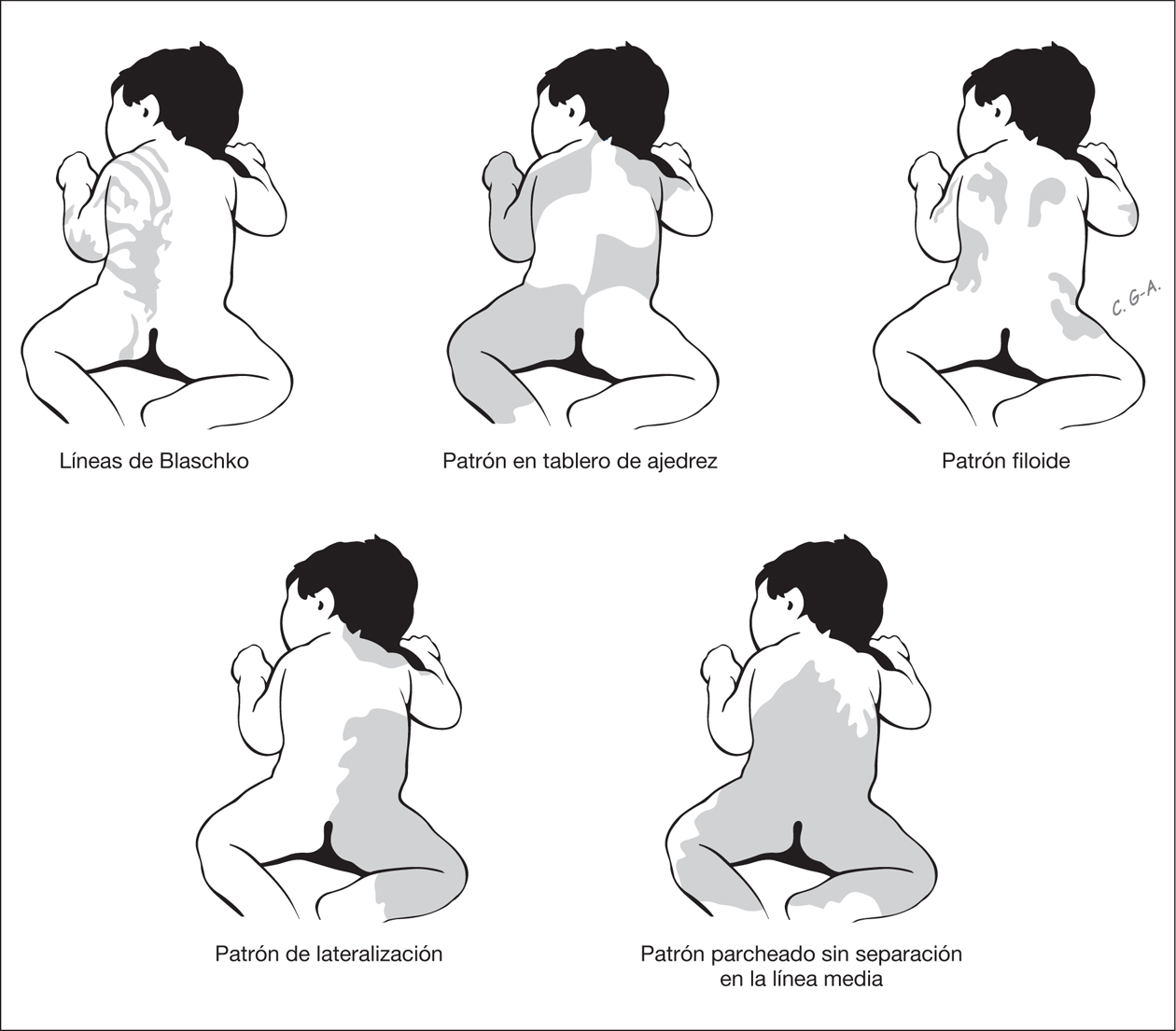
Figura 2. Patrones de alteraciones cutáneas que sugieren mosaicismo cutáneo.
Las líneas de Blaschko resultan de la proliferación transversal desde la línea primitiva de células precursoras de la epidermis y su migración durante la embriogénesis temprana de la piel siguiendo patrones específicos 3. Varios trastornos de la piel siguen estos patrones lineales, en los cuales las bandas de piel anormal, hipopigmentadas o hiperpigmentadas, representan la proliferación clonal de una población de células funcionalmente diferentes que portan la alteración en un gen expresado en la epidermis. Las bandas lineales muestran un patrón variable y pueden ser estrechas o anchas y su expresión en el neonato puede ser bastante tenue y pasar desapercibidas. Las alteraciones cutáneas que se distribuyen siguiendo las líneas de Blaschko se observan en entidades muy heterogéneas (tabla 1).

Las alteraciones cutáneas que siguen un patrón en tablero de ajedrez se distribuyen en grandes áreas bien delimitadas y con una clara delimitación de la línea media. Happle ha sugerido que quizá los nevos flamígeros observados en el síndrome de Klippel-Trénaunay-Weber pueden estar dispuestos siguiendo este patrón 3. La distribución siguiendo un patrón filoide se caracteriza por máculas hipopigmentadas rectangulares o en forma de hoja, un patrón que recuerda a ornamentos florales. Este patrón se observa en pacientes con trisomía 13 en mosaico, los cuales presentan ausencia de cuerpo calloso, sordera de conducción, trastorno cognitivo, coloboma retiniano y coroideo, defectos craneofaciales y alteraciones de los dedos. La distribución en forma de mancha grande que atraviesa la línea media es el patrón característico del nevo melanocítico gigante, si bien el mosaicismo no se ha demostrado aún en esta lesión. Por último, existe una distribución que sigue un patrón de lateralización, en la que la alteración cutánea afecta a la mitad del cuerpo sin pasar la línea media. Este patrón se observa en el síndrome CHILD (congenital hemidysplasia with icthyosiform erythrodermia and limb defects), que se caracteriza por hemidisplasia congénita, nevo ictiosiforme y defectos de las extremidades 3.
Lesiones cutáneas focales en la línea media del neuroeje
Estas lesiones pueden indicar la existencia disrafismo espinal oculto, que incluye lipomas intradurales, diastematomielia, mielocistoceles y médula anclada. Estas alteraciones pasan inadvertidas por la integridad de la cobertura cutánea sobre la lesión y por la ausencia habitual de expresión clínica neurológica durante el período neonatal. Las lesiones cutáneas asociadas con disrafismo espinal oculto son hipertricosis focal, lipoma, angioma, nevo y senos dermoides, así como el apéndice lumbosacro (fig. 3) 4. Los senos dermoides, pequeños orificios en la piel que se continúan por un estrecho conducto, se localizan en la línea media del neuroeje, y pueden observarse desde el occipucio hasta la región sacra, aunque en más del 70 % de los casos presentan localización lumbar o lumbosacra 5. En las fositas, a diferencia de los senos, no se asocia patología intradural. Mientras los senos dermoides se ubican siempre por encima del surco interglúteo y el tracto se orienta en dirección cefálica, las fositas se ubican en el surco interglúteo y se orientan en dirección recta o caudal 5,6. Los senos dermoides puede infectarse y producir meningitis, con frecuencia recurrente. La hipertricosis focal se asocia con frecuencia a diastematomielia. Una masa subcutánea focal es una alteración que puede indicar un meningocele o un lipoma subcutáneo intradural. Este último se presenta en casi el 26 % de los pacientes con disrafismo espinal oculto 7. Dos lesiones distintivas asociadas con disrafismo espinal oculto son el apéndice lumbosacro y la "quemadura de pitillo". La primera, el denominado rabo humano, se asocia con disrafismo espinal (60 %), lipoma (30 %) y médula anclada (26 %) 8. La denominada "quemadura de pitillo" es una placa delimitada de piel atrófica, de forma circular en la línea media y próxima a la unión lumbosacra 9.
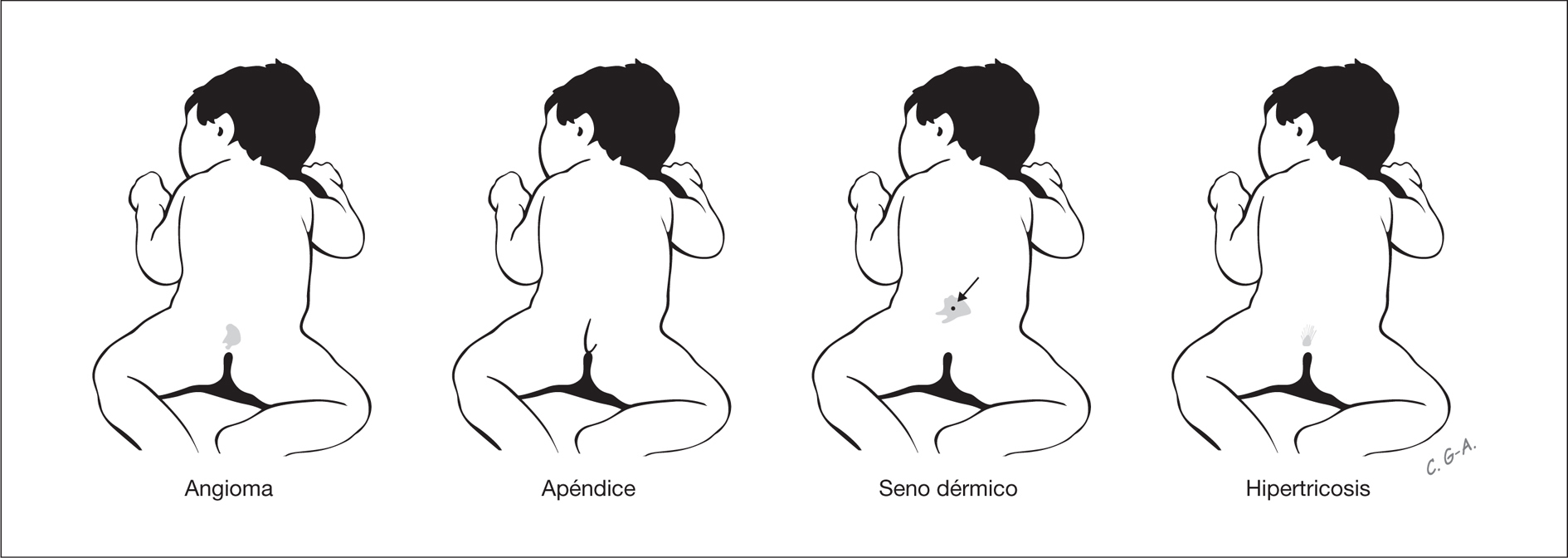
Figura 3. Lesiones cutáneas en la línea media del neuroeje que conllevan riesgo de disrafismo espinal oculto.
El disrafismo espinal oculto lumbar o lumbosacro cursa con alteración del cono medular y del filum terminal, estructuras que suelen estar fijadas por bandas fibrosas, lipoma o por tractos de los senos dermoides. La expresión clínica se caracteriza por dolor, déficit sensorial y motor en las extremidades inferiores, pies equinovaro, escoliosis y disfunción vesical e intestinal. Aunque la expresión clínica neurológica es rara en el recién nacido; ocasionalmente se observan alteraciones sensoriomotoras 4. La gran mayoría de los niños con defectos en la formación del tubo neural caudal presentan lesiones cutáneas y/o subcutáneas en el área lumbosacra 4-6. Reconocer estas alteraciones en la línea media del neuroeje como indicadores de potencial disrafismo espinal oculto facilita su diagnóstico precoz y en consecuencia la posibilidad de intervenciones quirúrgicas precoces que eviten el desarrollo de complicaciones y secuelas neurológicas. En estos recién nacidos debe evitarse la punción lumbar al existir riesgo de punción inadvertida de la médula espinal por estar el cono medular descendido. Tampoco debe introducirse sonda alguna para explorar la terminación del tracto del seno dermoide, ya que existe el riesgo de lesionar estructuras neurales, así como introducir bacterias y contaminantes de la piel 7.
Lesiones cutáneas hemorrágicas
Durante el nacimiento, la piel es expuesta a las fuerzas mecánicas, por lo que petequias, equimosis, abrasiones y laceraciones que aparecen en las primeras horas de vida pueden tener su origen en el parto e indicar un traumatismo obstétrico. Ante marcas rojas o abrasiones indicativas de parto por fórceps es obligado examinar la función del nervio facial para descartar paresia por traumatismo del nervio.
La presencia de lesiones cutáneas hemorrágicas generalizadas (petequias y equimosis) puede observarse en defectos congénitos de la coagulación y en las infecciones fetales sintomáticas por virus o protozoos que pueden producir alteraciones del SNC, en particular las infecciones por citomegalovirus o rubéola 10-13. Además de la púrpura o petequias, en la rubéola congénita se pueden observar pequeñas lesiones purpúricas debidas a eritropoyesis dérmica que dan lugar al denominado síndrome de blueberry muffin (pastel de arándanos).
Piel reticulada o marmórea
La presencia de una piel reticulada, que presenta aspecto de mármol con vetas azul-violeta, sugiere una cutis marmorata telangiectásica congénita (CMTC). En este cuadro las lesiones cutáneas suelen estar presentes desde el nacimiento, pueden ser localizadas o casi generalizadas y el patrón marmóreo se acentúa con el frío o el estrés. Durante las primeras semanas de vida, las lesiones parecen menos reticuladas y recuerdan a una malformación capilar. La CMTC se observa con relativa frecuencia en el síndrome de Down, en el de Lange y en la homocistinuria. Sin embargo, la mayoría de los casos son esporádicos y se cree que se trata de un mosaicismo cutáneo 3. Diversas anomalías pueden asociarse a la CMTC, entre estas la asimetría corporal (~ 43 %), anomalías neurológicas (~ 16 %) y oculares (~ 9 %) 14. Existe una forma específica de CMTC con macrocefalia al nacimiento (M-CMTC) a la que se puede asociar también macrosomía, hemihipertrofia, angiomas, piel hiperelástica y sindactilia 15. Una revisión de 75 casos de M-CMTC ha mostrado que el 75 % de estos pacientes presentan un nevo flamígero en el filtrum y/o del labio, por lo que esta alteración cutánea constituye una adecuada pista para su sospecha diagnóstica 16. El 61 % de los niños con M-CMTC desarrollan hidrocefalia posnatal y el 36 % presenta hemimegalencefalia 15,16.
Ausencia de piel-ulceración cutánea
La aplasia congénita del cutis (ACC) es un grupo heterogéneo de trastornos cutáneos que cursan con ausencia aislada o múltiple de la piel. Las lesiones son úlceras bien delimitadas, de forma oval o circular que se ubican con más frecuencia en el cuero cabelludo, principalmente en el vértex del occipucio, pero pueden presentarse en cualquier localización excepto en las palmas de las manos y las plantas de los pies 1,17. La superficie de la lesión puede parecer seca y curada o húmeda con exudado seroso y en ocasiones remedar una lesión hemorrágica. La lesión puede interesar a la epidermis y la dermis superior o puede extenderse a la dermis profunda, el tejido subcutáneo y en ocasiones cuando se ubica en el cuero cabelludo, al hueso subyacente. La gran mayoría, hasta el 70 %, son lesiones solitarias 17-19. Tras la curación, el área incluida en el defecto queda plana o deprimida, pero siempre sin pelo. Aunque la mayoría de las lesiones de ACC son esporádicas y no cursan con otras anomalías asociadas, estas lesiones pueden presentarse con otras anomalías congénitas de origen disruptivo o asociarse a diversos síndromes malformativos (tabla 2) 18,19. En ambas circunstancias pueden existir trastornos del SNC 20. Numerosas lesiones "en sacabocado" en el cuero cabelludo o en el remolino parietal en un recién nacido con retraso del crecimiento intrauterino y múltiples malformaciones congénitas sugiere una trisomía 13.

La ACC debe diferenciarse de la ausencia congénita localizada de la piel que se presenta sobre la cara anterior de la porción distal de las extremidades inferiores y el dorso de los pies, que junto con vesículas, ampollas mucocutáneas y alteraciones de las uñas constituye el síndrome de Bart, un probable subtipo de epidermólisis distrófica ampollosa a la que no se asocian alteraciones del desarrollo del SNC 21. Una rara entidad que cursa con manifestaciones cutáneas necróticas en una extremidad, generalmente el antebrazo, es la contractura isquémica congénita de Volkmann. Este es un síndrome compartimental excepcional, presente al nacimiento, que se caracteriza por necrosis muscular y parálisis del nervio que conduce a fibrosis muscular y contractura en flexión de los dedos. Las lesiones cutáneas son los hallazgos más distintivos al nacimiento y con frecuencia se observa una placa necrótica azulada, bien delimitada, de bordes irregulares, con afectación circunferencial de la extremidad y de extensión variable. También puede observarse edema, discoloración o ampollas 22.
Trastornos de la pigmentación
En general, la piel del bebé está menos pigmentada que la del niño y existe una amplia variabilidad en el grado y distribución de la pigmentación normal. Las alteraciones de la pigmentación son un buen marcador para el diagnóstico de trastornos neurocutáneos, neurocristopatías y síndromes genéticos específicos. En niños con epilepsia idiopática se ha observado una mayor incidencia de máculas hipopigmentadas y de manchas "café con leche" que en una población control de niños sanos, lo cual indica que estos trastornos constituyen un factor de riesgo de epilepsia 23.
Hipopigmentación difusa
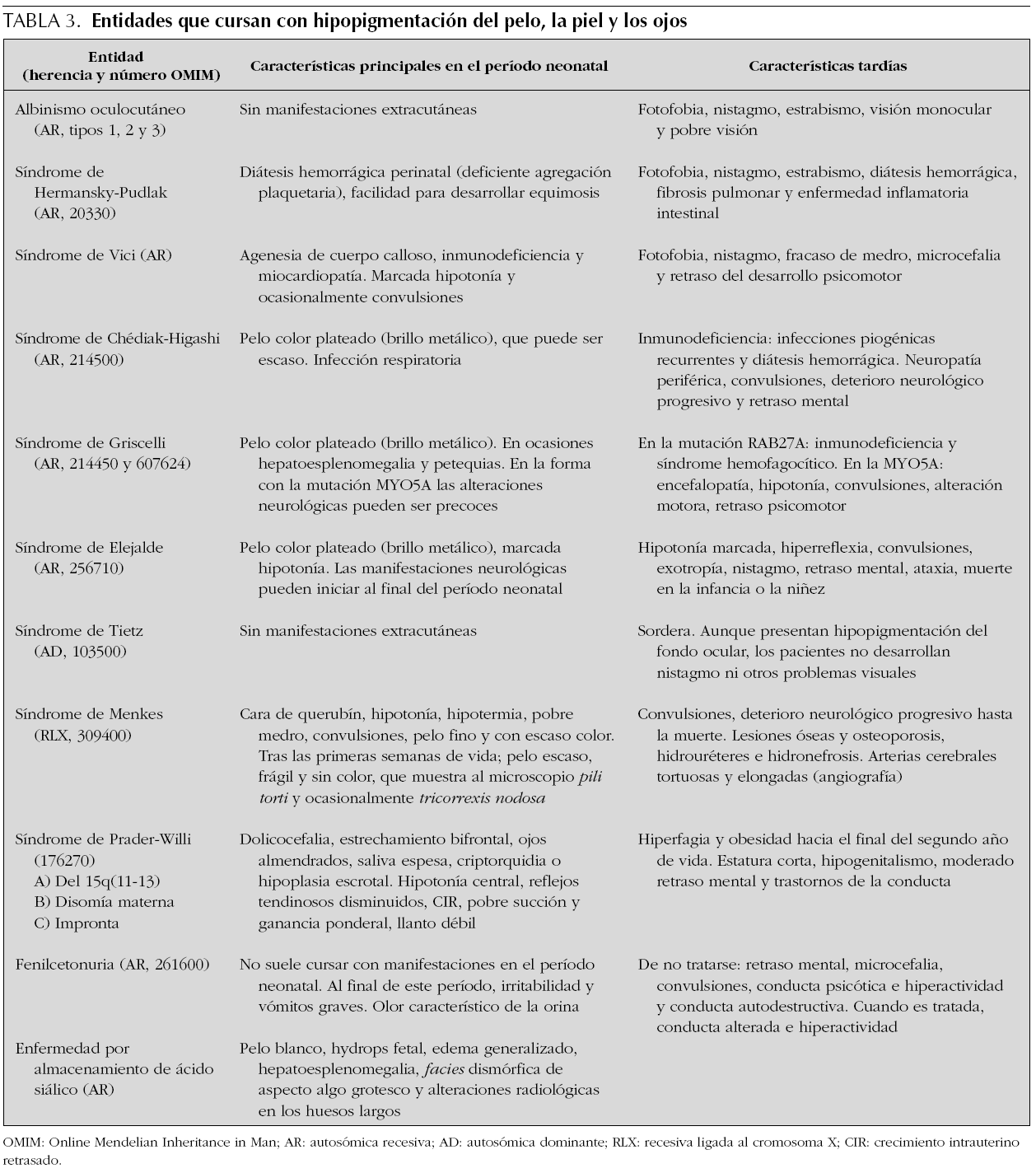
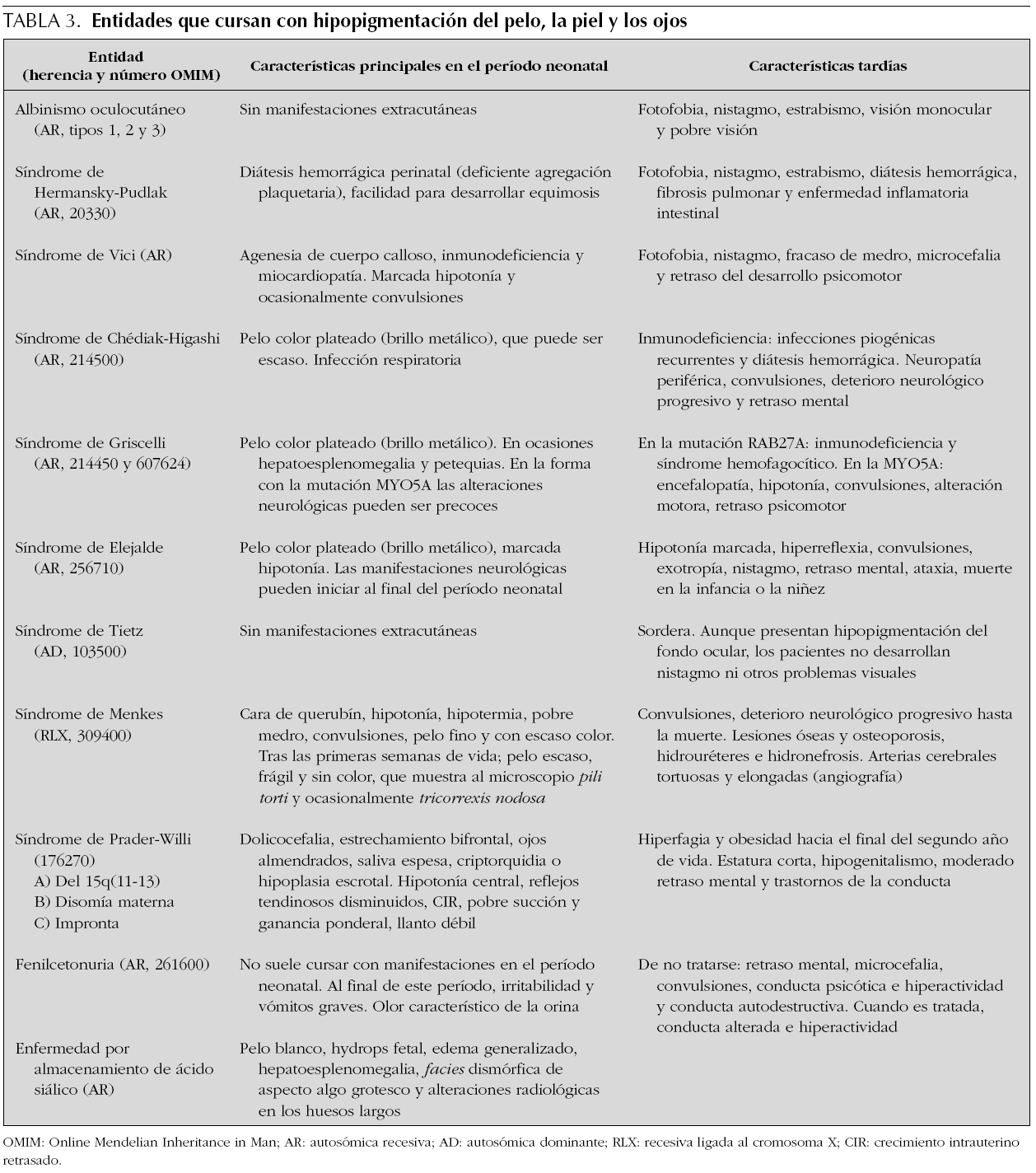
Los trastornos hereditarios de la pigmentación debidos a una alteración en la síntesis o distribución de melanina en la piel, el pelo y los ojos se agrupan bajo el término albinismo, y este incluye principalmente los siguientes trastornos: el albinismo oculocutáneo, el albinismo ocular, el síndrome de Chédiak-Higashi y el síndrome de Hermansky-Pudlak 24. Además de estas entidades, una gran diversidad de síndromes genéticos cursa con hipopigmentación cutánea y del iris (tabla 3).
Hipopigmentación parcheada
Cuando la hipopigmentación es parcheada la forma y la distribución de las lesiones es de gran ayuda para establecer la sospecha diagnóstica. Lesiones en forma de hoja de fresno de distribución proximal son características de la esclerosis tuberosa, mientras que lesiones hipopigmentadas lineales en el tronco o las extremidades sugiere una hipomelanosis de Ito y áreas amelanóticas de forma triangular sobre la frente pueden sugerir un síndrome de Waardenburg (fig. 4).
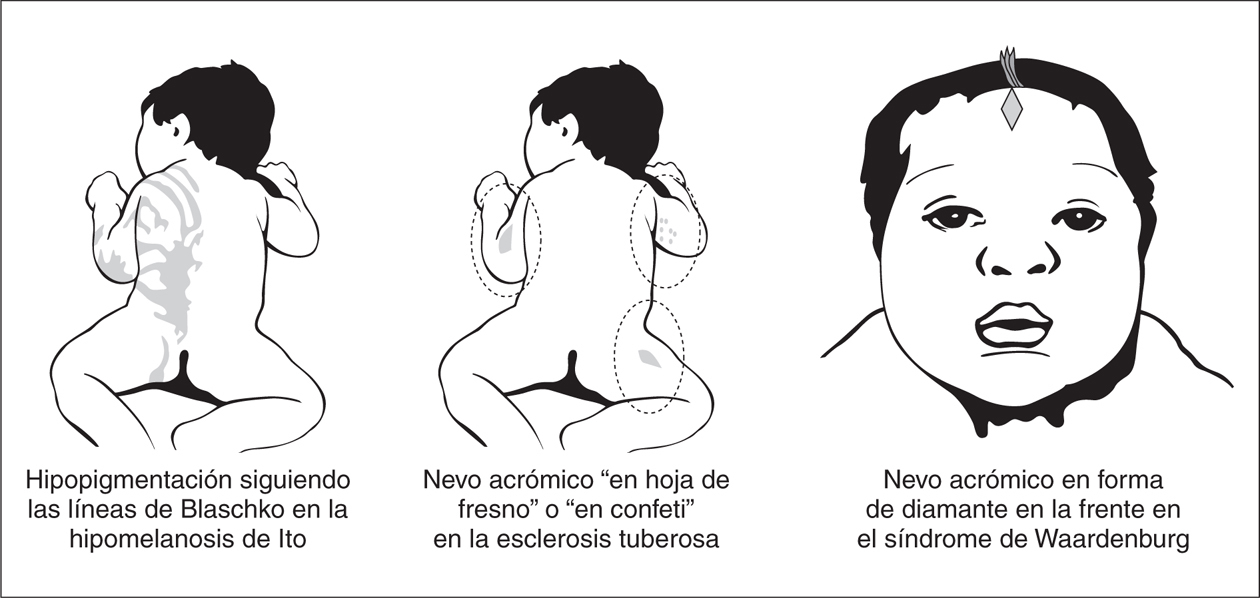
Figura 4. Entidades con hipopigmentación parcheada.
La hipomelanosis de Ito se caracteriza por lesiones hipopigmentadas asociadas a alteraciones neurológicas y otras anomalías extraneurológicas 25-28. Las lesiones siguen una distribución lineal o segmentaria y se observan con frecuencia en el tronco, ocasionalmente en las extremidades y rara vez en la cara de forma unilateral o bilateral, pero nunca en las palmas, las plantas, el cuero cabelludo o en las membranas mucosas 25. La distribución varía de tiras lineales siguiendo las líneas de Blaschko a una distribución en remolinos o placas hipopigmentadas con márgenes irregulares. Las lesiones tienen un fuerte predominio unilateral, rara vez son bilaterales y en ocasiones pueden asociarse otras lesiones cutáneas como manchas "café con leche", nevo de Ota y nevo angiomatoso o marmóreo 25-28. Las lesiones pueden estar presentes en el período neonatal; en una amplia serie de casos, las lesiones cutáneas se observaron en el 64 % de los pacientes 27. Cuando esta hipopigmentación no se asocia con alteraciones neurológicas o de otros sistemas, se denomina naevus depigmentosus. La hipomelanosis de Ito asocia con frecuencia anomalías del SNC (alteraciones de la sustancia blanca, paquigiria, heterotopias neuronales, displasia de los ganglios basales, atrofia del cerebelo y en ocasiones hemimegalencefalia), y/o del periférico, así como anomalías oculares (microftalmía, opacidades corneales, atrofia coroidea) y anomalías esqueléticas (hemihipertrofia, escoliosis), del pelo, dientes y uñas 25-28. En el presente la hipomelanosis de Ito no se considera una entidad específica, sino un grupo heterogéneo de procesos neurocutáneos con diferentes formas de mosaicismo cutáneo 29,30.
Lesión hipomelanótica triangular o en forma de diamante sobre la frente
Esta lesión característica del síndrome de Waardenburg, un síndrome pigmentario-auditivo de herencia dominante con penetrancia variable, puede estar presente desde el nacimiento. Otras alteraciones, como el mechón de pelo blanco, las áreas de vitíligo en la piel, la heterocromía de iris y la sordera se manifestarán en etapas más tardías 31. El síndrome de Waardenburg debe diferenciarse del piebaldismo, un cuadro que cursa también con una mancha amelanótica bien delimitada en la frente junto con otras áreas de vitíligo en la piel. Sin embargo, el piebaldismo no cursa con sordera ni con las demás características del síndrome de Waardenburg.
Lesión hipomelanótica en forma de hoja de fresno
Máculas hipomelanóticas se observan con frecuencia en niños con esclerosis tuberosa. Estas lesiones cutáneas son habitualmente las únicas anomalías cutáneas de esclerosis tuberosa presentes al nacimiento 1. Las lesiones cutáneas características tienen forma de hoja de fresno (forma oval larga y estrecha) o son redondeadas y segmentarias, recordando a confeti. Estas máculas hipopigmentadas se observan mejor en recién nacidos con elevada pigmentación racial, pero pueden no ser visibles en aquellos de piel clara, y se hace entonces necesario el examen con lámpara de Word. Los fibromas periungueales, los angiofibromas faciales y las placas de "shagreen" aparecen más tarde, rara vez antes de los 2 años. Sin embargo, los estudios de neuroimagen pueden mostrar nódulos subependimarios en la sustancia blanca desde el período neonatal 32.
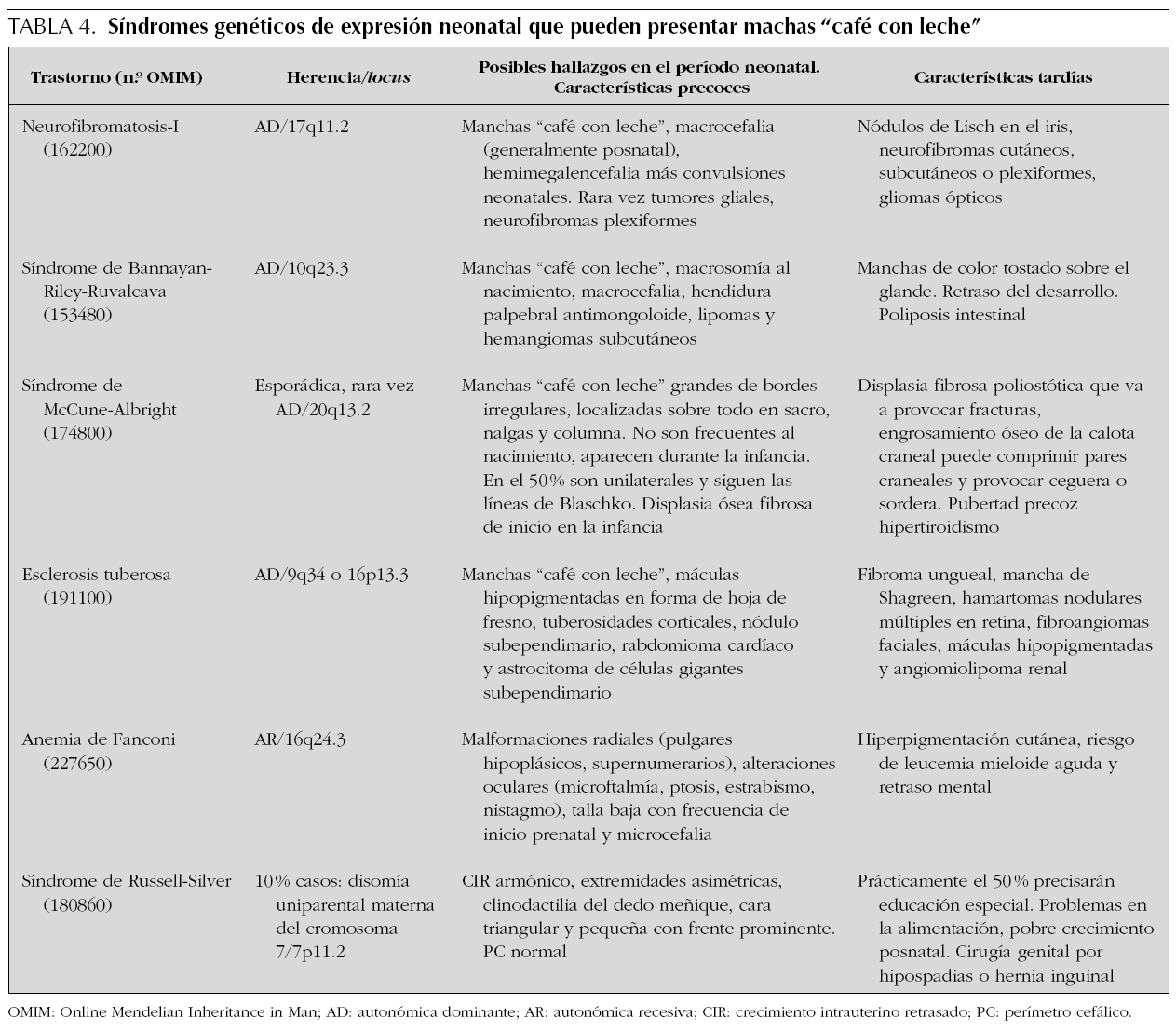
Manchas "café con leche" y áreas hiperpigmentadas
Son máculas bien delimitadas, redondas u ovales de pigmentación uniforme. Su color varía entre castaño claro u oscuro y el borde está claramente delimitado, pero puede ser liso o irregular. La importancia clínica de las manchas "café con leche" se relaciona con su posible asociación con trastornos genéticos con expresión multiorgánica, los más frecuentes con afectación del SNC (tabla 4). Las manchas "café con leche" aisladas son relativamente frecuentes, pero si son múltiples puede indicar la presencia de un trastorno genético. La frecuencia y cantidad de las manchas "café con leche" varían en la población de acuerdo con el origen étnico y la edad. Aunque son más frecuentes en niños prepúberes o púberes pueden estar presentes desde el nacimiento. En un estudio que incluyó a más de 4.500 recién nacidos, se observaron manchas "café con leche" en el 0,3 % de los caucásicos y en el 18 % de los afroamericanos. Mientras que ninguno de los caucásicos tuvo más de una mancha "café con leche", el 6,3 % de los afroamericanos presentaron dos o más33. Existen pocos estudios longitudinales en lactantes con varias manchas "café con leche" dirigidos a examinar el riesgo de desarrollar posteriormente trastornos multiorgánicos o de tener una enfermedad genética subyacente. Por otra parte, se ha referido que alrededor del 20 % de los pacientes chinos con neurofibromatosis tipo 1 (NF1) presentan este tipo de manchas desde el nacimiento 34. Aproximadamente el 40-50 % de los recién nacidos con más de 5 manchas "café con leche" de más de 5 mm desarrollarán neurofibromatosis 35-37. Por ello, la presencia en un recién nacido de más de 5 manchas de 5 mm obliga a buscar otras características asociadas a estos síndromes, así como a realizar un detenido examen de los familiares dada la herencia autosómica dominante de la mayoría.
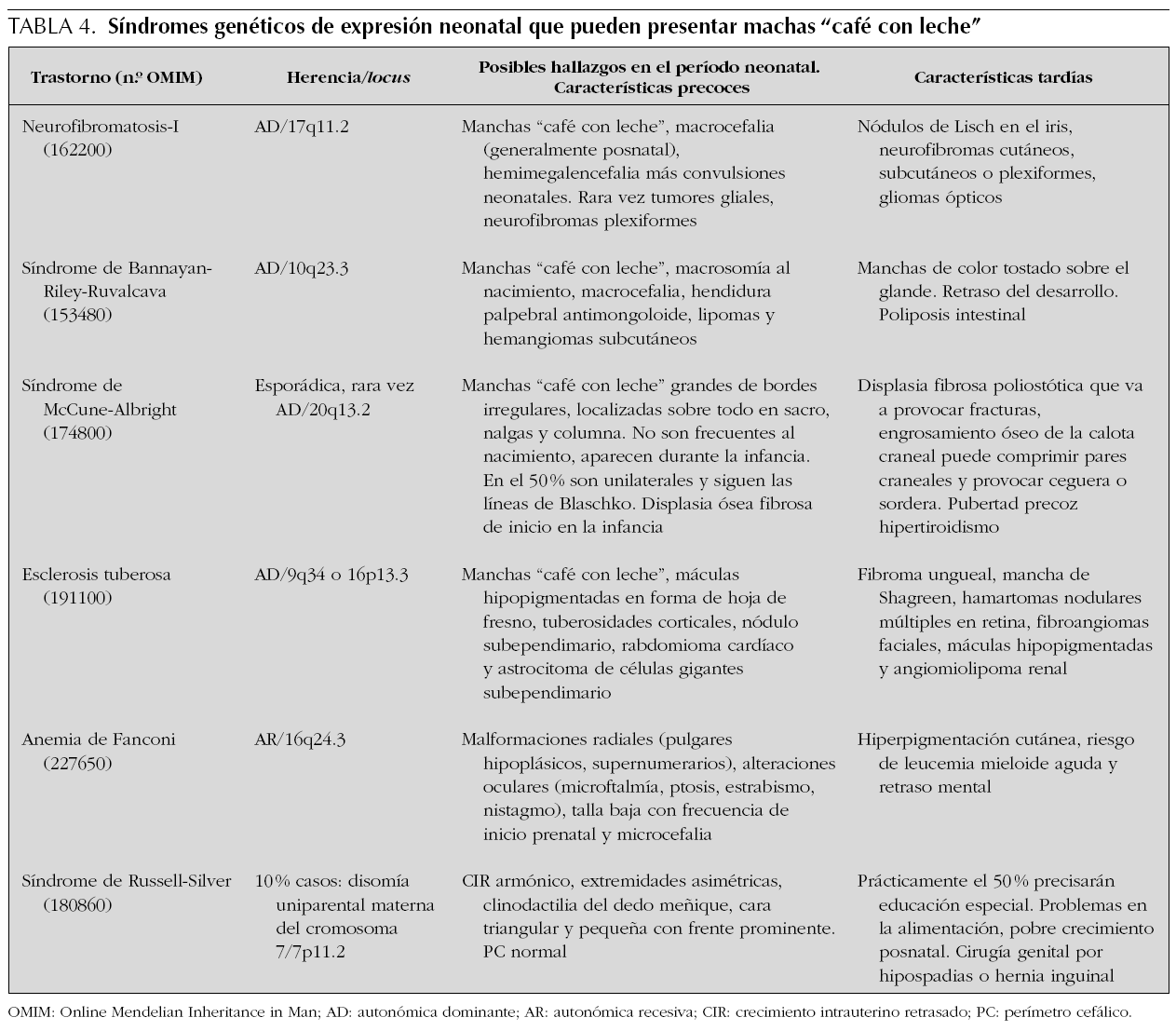
Nevos congénitos
En un sentido amplio se usa el término nevo para describir cualquier lesión pigmentada. Algunos nevos congénitos tienen una especial trascendencia por ser expresión de síndromes neurocutáneos. Los nevos epidérmicos son lesiones hamartomatosas caracterizadas por hiperplasia de elementos de la epidermis que derivan de las células ectodérmicas y se clasifican de acuerdo al componente principal que puede ser sebáceo, apocrino, ecrino, folicular o queratinocítico. El nevo sebáceo de Jadassohn es el tipo de nevo epidérmico más común y está presente hasta en el 0,3 % de los recién nacidos 33. Estos nevos suelen ser únicos y se localizan preferentemente en el cuero cabelludo o la cara como áreas de 1 a 10 cm con forma oval, redondeada o lineal, sin pelo o vello. La superficie del nevo está ligeramente elevada, de color amarillo o naranja, con pequeñas pápulas y de superficie encerada o aterciopelada al tacto (fig. 5). Generalmente, al inicio de la pubertad, la lesión se volverá más verrugosa y nodular. El nevo epidérmico lineal verrugoso es el segundo tipo más frecuente, y se observa en forma de mancha poco coloreada y ligeramente escamosa que nunca atraviesa la línea media. Con la maduración, pasa a ser una lesión lineal, engrosada, verrugosa e hiperpigmentada. Los nevos epidérmicos se ubican preferentemente en la cara, el cuero cabelludo o en el cuello pero en ocasiones, con la maduración, estas lesiones pueden distribuirse en áreas extensas del cuerpo a lo largo de las líneas de Blaschko.
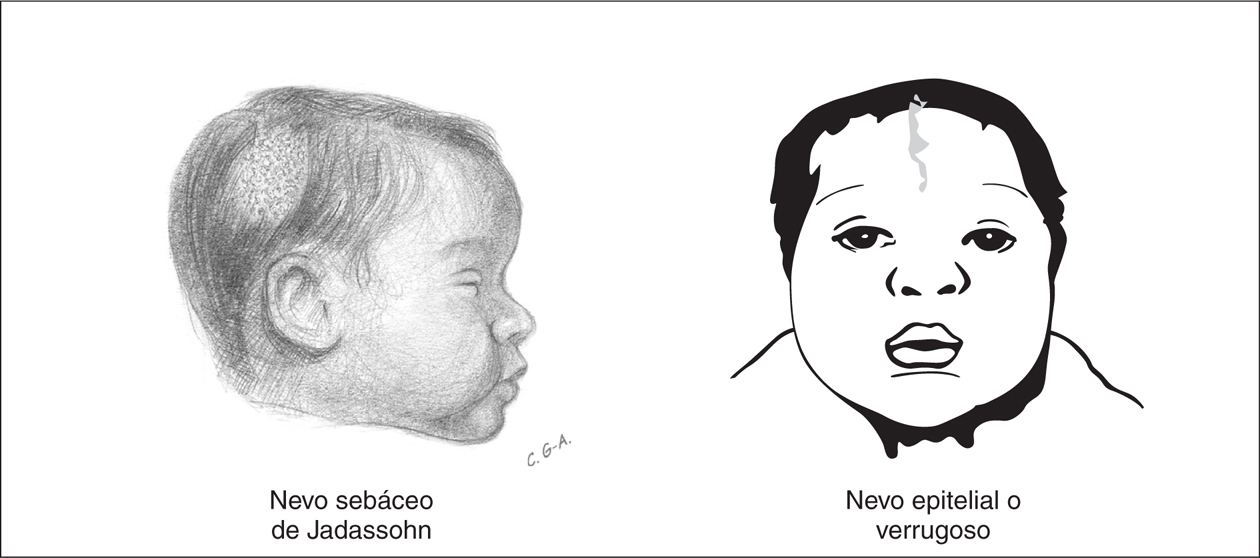
Figura 5.Nevos epidérmicos que, asociados a otras anomalías orgánicas, particularmente del SNC, constituyen el síndrome del nevo epidérmico.
Los nevos epidérmicos sebáceos tienen potencial para malignizarse; se señala un riesgo de un 15-20 % de epitelioma de células basales. Además, pueden asociarse a otras anomalías orgánicas, en particular del SNC, constituyendo el denominado síndrome del nevo epidérmico. Sin embargo, este no es una entidad única y según el tipo de nevo epidérmico pueden reconocerse diversos tipos de síndrome del nevo epidérmico 38,39. Este puede cursar con un amplio espectro de alteraciones neurológicas, oftalmológicas, esqueléticas, cardiovasculares y urogenitales. Los tipos de presentación neonatal más frecuentes son el síndrome del nevo epidérmico lineal y el síndrome del nevo sebáceo lineal. Las alteraciones neurológicas no suelen expresarse en el período neonatal y lo hacen a los pocos meses de vida. La principal alteración neurológica en el período neonatal son las convulsiones y posteriormente la epilepsia, el retraso mental, la hemiparesia y la parálisis de nervios craneales 40-42. La prevalencia de la afectación del SNC parece ser más elevada cuando el nevo epidérmico afecta a la cabeza o la cara. Anomalías estructurales del SNC están presentes en más del 70 % de los casos; ventriculomegalia, hemimegalencefalia, hemimegacráneo, hidrocefalia, lisencefalia unilateral e hipoplasia cerebelosa. La proporción de nevos sebáceos de Jadassohn que se dan en el contexto de un síndrome del nevo epidérmico se desconoce, así como el riesgo exacto de malignización posterior 1.
Se ha identificado un subgrupo de niños con síndrome del nevo epidérmico que presentan nevo epidérmico facial, hemimegalencefalia ipsolateral, malformación de la circunvolución, retraso mental, convulsiones y que con frecuencia desarrollan hemihipertrofia facial ipsolateral 41,42. Las anomalías estructurales que presentan son: hemimegalencefalia, ventriculomegalia y aumento de la intensidad de señal en T2 en la sustancia blanca del hemisferio afectado 41,42. La presencia de un nevo epidérmico en un recién nacido, en particular en la cabeza o en área mediofacial, obliga a realizar al menos una evaluación ultrasonográfica cerebral.
Dos entidades incluidas como tipos de "síndrome del nevo epidérmico" son el síndrome de Proteus (v. más adelante) y el síndrome CHILD, acrónimo que señala una entidad ligada al sexo caracterizada por hemidisplasia congénita, nevo ictiosiforme y defectos en los miembros con un patrón de lateralización muy acusado, que puede presentar anomalías viscerales y del SNC ipsolaterales como la hipoplasia cerebral unilateral.
El nevo melanocítico congénito es un nevo hiperpigmentado de bordes bien definidos que se debe a un aumento excesivo del número de melanocitos en la capa basal de la epidermis y en la dermis 1. Estos nevos pueden ser pilosos y se distribuyen con frecuencia en el tronco inferior o en la parte baja o superior de la espalda y las nalgas. Las lesiones pueden ser planas, elevadas, verrugosas o nodulares y pueden tener diversas sombras marrón, azul o negro. Algunos nevos, en particular los gigantes, pueden asociar múltiples nevos (tres o más) satélites. Entre el 1 y el 6 % de los recién nacidos presentan nevos melanocíticos congénitos al nacimiento y según el diámetro de mayor tamaño se clasifican arbitrariamente como: gigantes (> 20 cm), intermedios (entre 1,5 y 19,9 cm) y pequeños (< 1,5 cm) 43. Los nevos melanocíticos congénitos no tienen segregación familiar y los gigantes ocurren en aproximadamente 1:20.000 recién nacidos 44. Los nevos melanocíticos congénitos, en particular los gigantes, representan un aumento del riesgo de desarrollar melanomas y también de melanosis neurocutánea: invasión y proliferación melanocítica benigna o maligna en el SNC, tanto en las leptomeninges como en determinadas regiones del parénquima cerebral. Un reciente estudio de 160 niños con nevo melanocítico congénito gigante, seguidos 5 años por término medio, mostró que estos pacientes tienen un riesgo acumulado de presentar melanoma del 2,3 % (intervalo de confianza [IC] del 95 %, 0,8-6,6) y de desarrollar melanosis neurocutánea del 2,5 % (IC 95 %, 0,8-7,2) 45. Por lo general, los niños que desarrollan melanosis neurocutánea presentan síntomas neurológicos antes de los 3 años y la mayoría fallecen en los primeros 3 años tras iniciarse la sintomatología neurológica 45. Las convulsiones y la hidrocefalia son las manifestaciones más frecuentes, esta última probablemente debido a bloqueo de la resorción o de la circulación del LCR. Además de los melanomas y la melanosis neurocutánea, los nevos melanocíticos congénitos de más de 2 cm de diámetro se puede asociar una gran diversidad de anomalías estructurales no melanocíticas del SNC. Se recomienda realizar estudios tempranos de neuroimagen para examinar no sólo la posibilidad de melanosis intracraneal, sino también la posible presencia de alteraciones estructurales 46.
Hemangionas y malformaciones vasculares
Los hemangiomas son lesiones vasculares benignas de la piel y otros tejidos que poseen espacios recubiertos por endotelio y una estroma fibrosa o fibromucinosa 47. Mulliken y Glowacki 48 han propuesto diferenciar entre hemangiomas y malformaciones vasculares de acuerdo con las características clínicas, el curso clínico y particularmente en función de los hallazgos histológicos. Según sus criterios, los hemangiomas son verdaderos tumores vasculares (lesiones proliferativas) que crecen rápidamente y muestran un recambio aumentado de células endoteliales y un incremento en el número de células mastoides. Los hemangiomas por lo general no se observan inmediatamente tras el nacimiento, muestran un rápido crecimiento posnatal seguido por una lenta involución. Están presentes hasta en el 10 % de los lactantes, pero rara vez se asocian a malformaciones sistémicas y/o a anomalías del desarrollo del SNC. Las malformaciones vasculares son errores innatos en la morfogénesis vascular que muestran un recambio normal de células endoteliales y un número normal de células mastoides. Están presentes al nacimiento, aunque pueden ser poco evidentes, muestran un crecimiento proporcionado con el crecimiento del lactante y no disminuyen de tamaño por involución 48. En general, las anomalías vasculares que asocian con más frecuencia anomalías del SNC son los hemangiomas capilares y los nevos flamígeros.
Hemangioma capilar o hemangioma en frambuesa
Son tumores vasculares rojo brillantes, elevados, bien delimitados, blandos y compresibles. Con frecuencia son lobulados, varían ampliamente en tamaño y localización y pueden localizarse en cualquier parte del cuerpo. Por lo general no están presentes al nacimiento, aparecen entre la tercera y la quinta semana de vida y pueden ser precedidos por palidez de la piel afectada y poco después por una mancha eritematosa o discreta telangiectasia. Son más prevalentes en mujeres y en los prematuros 49. Estos hemangiomas tienen una fase de rápido crecimiento posnatal durante los primeros meses, seguida por una fase de lenta regresión a partir del año, alcanzando la resolución completa en cerca del 70 % de los niños para los 7 años de edad y en el 90 % para los 10 años 50. La presencia de múltiples hemangiomas cutáneos sin afectación visceral se denomina hemangiomatosis neonatal benigna, pero cuando existe afectación visceral (hígado, cerebro) se habla de hemangiomatosis neonatal diseminada. Hemangiomas capilares múltiples en la cara de forma unilateral o bilateral, grandes con conducta agresiva (crecimiento activo) y a veces con distribución trigeminal, se asocian con malformación de Dandy-Walker u otras malformaciones en fosa posterior, así como con anomalías cardíacas, arteriales y oculares 51,52. Este conjunto de anomalías asociadas a hemangioma facial se ha agrupado bajo el acrónimo PHACE/PHACES 53,54. El espectro de anomalías asociadas puede ser más amplio que el incluido en dicho acrónimo y se ha propuesto denominar a esta entidad síndrome de Pascual-Castroviejo II 55. La asociación de hemangiomas capilares con macrocefalia y lipomas sugiere el síndrome de Bannayan-Zonana. Un cuadro muy raro en el cual aproximadamente la mitad de los pacientes presenta malformación vascular arteriovenosa que afecta al paladar, la mucosa oral, maxilar y mandíbula es el síndrome de Wyburn-Mason o angiomatosis retinocefálica. Este trastorno cursa con la coexistencia de angioma tortuoso en el fondo ocular y malformación arteriovenosa intracraneal.
Nevo flamígero o "en vino de Oporto"
Esta alteración vascular se presenta como una lesión no elevada de color rojo oscuro o púrpura con bordes irregulares en menos del 1 % de los niños 56. En el momento del nacimiento son a menudo lesiones maculares de color rosa pálido que, con el tiempo, progresan hasta adquirir el color rojo oscuro. Esta anomalía vascular congénita no tiende a resolverse con la edad y es el nevo vascular que asocia con más frecuencia malformaciones vasculares de otros tejidos, sobre todo en los territorios subyacentes al nevo. Las lesiones tienden a crecer, engrosarse o adquirir un carácter nodular con la edad. Una mancha "en vino de Oporto" en el territorio del nervio trigémino crea preocupación por cuanto estos nevos vasculares pueden asociarse con anomalías de los vasos coroideos en el ojo, de los vasos meníngeos y de los vasos superficiales del cerebro; el síndrome de Sturge-Weber. Las anomalías vasculares en este síndrome pueden producir glaucoma y lesiones cerebrales corticales expresadas por convulsiones y otros síntomas neurológicos como hemiparesia y hemianopsia. Aunque las manifestaciones neurológicas no se presentan por lo general en el período neonatal, las alteraciones del parénquima cerebral pueden estar presentes desde el nacimiento 57,58. El patrón de distribución del nevo "en vino de Oporto" predice la probabilidad de afectación de los vasos coroideos y leptomeníngeos. Aproximadamente el 8 % de los nevos vasculares que afectan al territorio del trigémino presentan complicaciones oculares y/o del SNC y el riesgo varía de acuerdo con la distribución trigeminal del nevo "en vino de Oporto" 59,60. La afectación completa del párpado superior e inferior (V1) conlleva un alto riesgo (≈ 40 %), mientras que si el nevo está limitado al párpado superior (afectación parcial de V1) el riesgo es muy bajo y una distribución limitada a los dermatomas V2 y V3 no conlleva riesgo. Cuando la distribución es amplia, de V1 a V3, casi la mitad de los niños tendrán un síndrome de Sturge-Weber y casi el 25 % de los niños con nevo vascular bilateral. No parece existir relación entre el tamaño de nevo y la alteración clínica neurológica que presentará el niño.
Además de en el síndrome de Sturge-Weber, nevos flamígeros prominentes en la cara, pero en la frente y sobre la glabela, se observan en el síndrome de Beckwith-Wiedemann, de Rubinstein-Taybi y en el síndrome de Roberts. En la M-CMTC el nevo flamígero se localiza en el filtro y/o el labio superior 15.
Hiperqueratosis
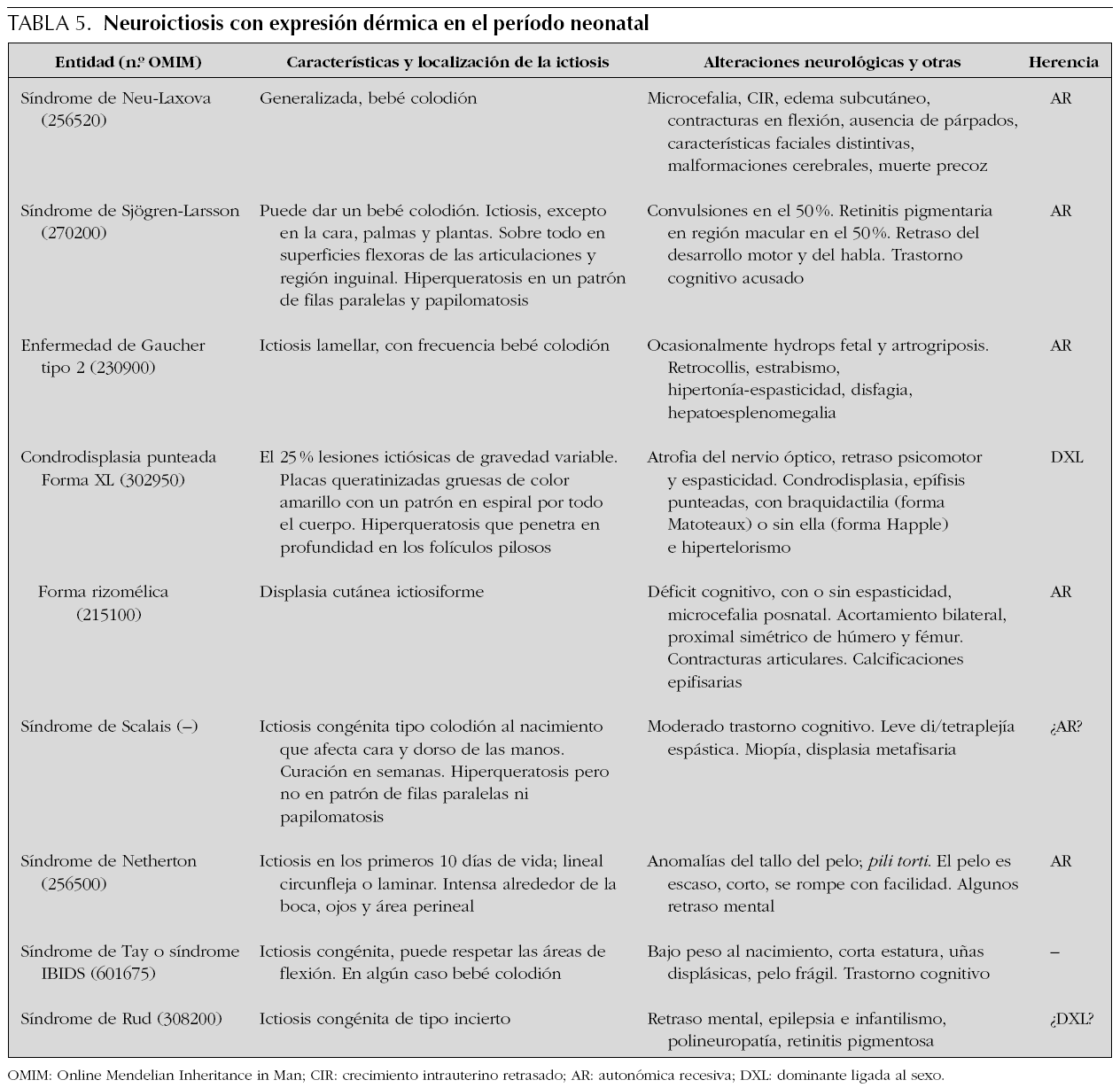
Las ictiosis son trastornos de la queratinización de la piel y constituyen un grupo heterogéneo de trastornos hereditarios. La ictiosis se puede presentar como un hallazgo cutáneo aislado o ser parte de un diverso número de síndromes y se caracteriza por la formación de escamas en la capa córnea de la piel. Las ictiosis difieren en su patrón de escamas, histopatología, genética y bioquímica, así como en los hallazgos asociados. Las formas más frecuentes de ictiosis con expresión neonatal no asocian afectación del SNC, pero algunas formas cursan con afectación neurológica durante la niñez y por ello se las denomina colectivamente como neuroictiosis 61. El prototipo de estas es el síndrome de Sjögren-Larsson, que se caracteriza por ictiosis congénita de curso benigno, di/tetraplejía espástica y retraso mental 62. Algunas neuroictiosis pueden presentarse como un bebé colodión al nacimiento; piel rígida, gruesa y tensa que parece un pergamino oleoso y que se agrieta a través de las flexuras. Las principales neuroictiosis con sintomatología dermatológica desde el período neonatal y alteración neurológica durante este período o posteriormente se muestran en la tabla 5.
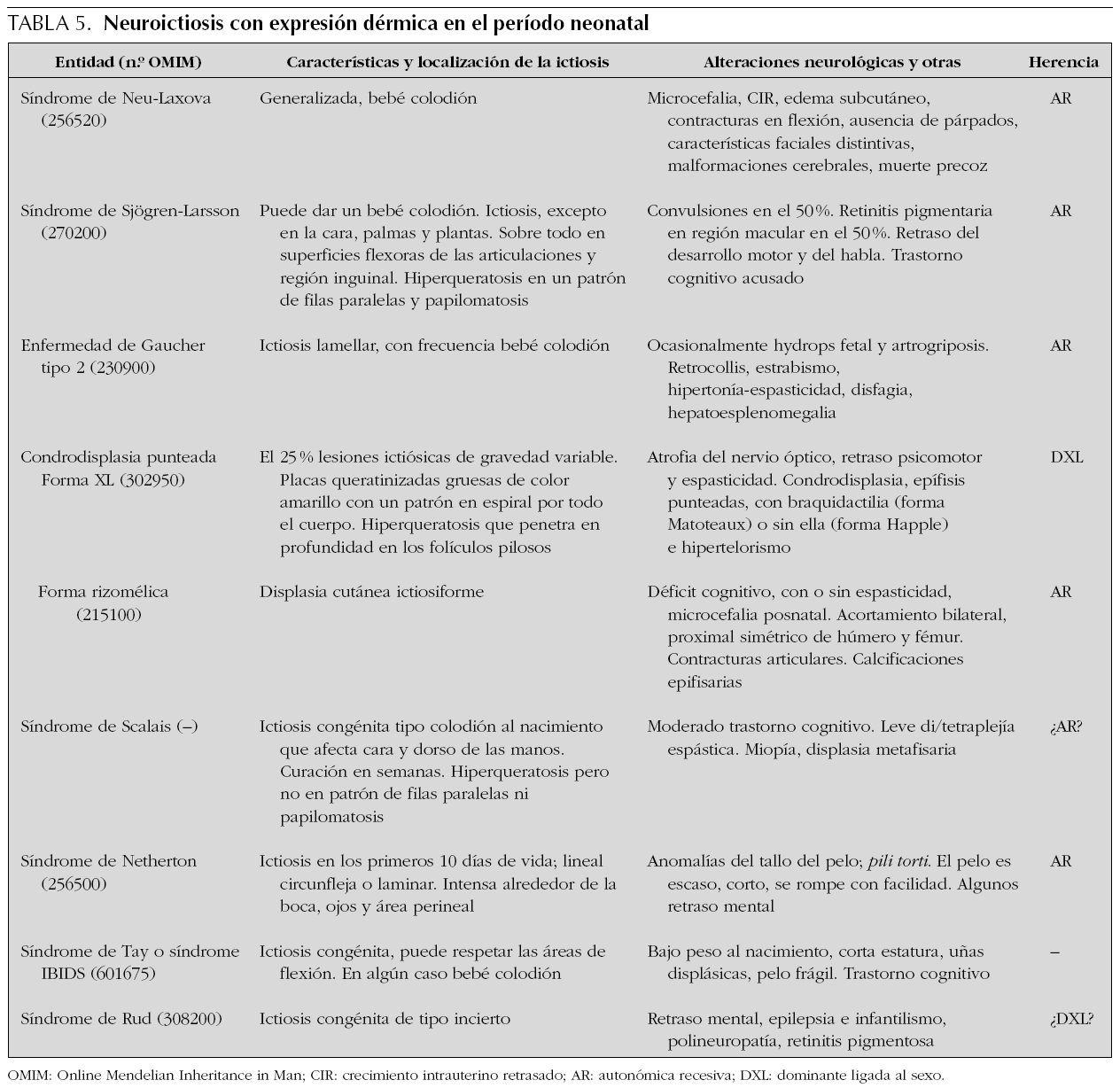
Lesiones vesiculosas
La presencia de vesículas en la piel de un recién nacido entre el final de la primera semana de vida y la tercera semana plantea siempre preocupación ante la posibilidad de que el bebé presente una infección perinatal por el virus herpes simple. Las tres formas de enfermedad herpética pueden presentar vesículas cutáneas; se observan en el 83 % de los recién nacidos con la forma localizada en piel, ojos y boca, en el 68 % de aquellos con afectación del SNC y en un 61 % de los pacientes con la forma diseminada 63. Cuando las vesículas aparecen en las primeras 24 h de vida sugieren una adquisición de la infección intrauterina 63. Las lesiones iniciales son máculas que en 48 h se transforman en vesículas, que se ubican preferentemente en las zonas de inoculación; las áreas de presentación (vértex o nalgas) o donde se alojaron los electrodos de monitorización 10. Las vesículas se distribuyen en grupos y tienen una base eritematosa. El máximo beneficio de la terapia antiviral se consigue cuando el tratamiento se inicia antes de la diseminación del virus en el organismo o antes de su replicación en el SNC 64. Por ello, la presencia de vesículas debe ser considerada una infección por herpes simple hasta que no se demuestre lo contrario 10. Pueden observarse vesículas pustulosas indistinguibles de las del herpes simple cuando se adquiere el virus varicela-zóster (VVZ) desde la madre justo antes de nacer. En estos casos, por lo general, las vesículas están distribuidas de forma más amplia que en el herpes 1.
En el período neonatal, en la fase eritematosa vesiculosa de la incontinencia pigmentaria aparecen lesiones vesiculares. Se trata de una enfermedad dominante ligada al sexo, letal en varones y que afecta casi de manera exclusiva a mujeres, que cursa con alteración de la piel, del SNC y de otros derivados ectodérmicos 1. La expresión cutánea de esta enfermedad tiene un curso trifásico, con un estadio eritematoso vesicular presente durante las primeras semanas de vida, en el cual las lesiones presentan una distribución lineal o agrupada. El líquido de las vesículas muestra un predominio de eosinófilos y la eosinofilia puede estar presente en la sangre periférica. Las lesiones pueden no ser aparentes tras el nacimiento, pero se harán progresivamente manifiestas durante las primeras semanas de vida con aparición de estrías lineales eritematosas y placas de vesículas en el cuero cabelludo, el tronco y las extremidades. Posteriormente tiene lugar un estadio verrugoso que se resuelve dejando áreas de atrofia y tiras de piel hipopigmentada. Hacia los 3-6 meses de vida se desarrolla el estadio hiperpigmentado, momento en el que se observan las características lesiones hiperpigmentadas con un patrón marmóreo que sigue las líneas de Blaschko. Más del 30 % de los niños con esta enfermedad presentan alteraciones neurológicas, incluidos trastornos cognitivos, convulsiones, espasticidad, convulsiones y microcefalia progresiva posnatal. También son frecuentes los trastornos oculares: estrabismo, nistagmo, ceguera, cataratas, atrofia óptica y ceguera. Las lesiones iniciales se confunden con herpes simple, impétigo ampolloso o mastocitosis. Sin embargo, la distribución lineal es exclusivamente característica de la incontinencia pigmentaria.
Lesiones cutáneas cicatriciales
La presencia en un recién nacido de lesiones cicatriciales que siguen la distribución de los dermatomas, tienen carácter serpenteante (en zigzag) y están deprimidas sugiere una infección del feto por varicela entre las 8 y las 20 semanas de gestación. Las extremidades en las que asientan estas cicatrices pueden estar atróficas, con musculatura hipoplásica y contracturas articulares. Prácticamente todos los casos cursan también con trastornos del SNC y oculares 10,65. Además de la hipoplasia, la paresia y la debilidad de las extremidades afectadas, estos recién nacidos pueden presentar dificultad de la deglución, microcefalia grave, convulsiones y atrofia cerebral. Las anomalías oculares son un hallazgo prácticamente universal; cataratas, coroidorretinitis, microftalmía, atrofia óptica o síndrome de Horner. El diagnóstico de fetopatía por VVZ se basa fundamentalmente en los antecedentes de infección materna por varicela durante la gestación y, en menor medida, en los estudios serológicos 10,65.
Lesiones asimétricas al nacimiento que parecen cicatrices y que corresponden a áreas de aplasia o hipoplasia dérmica pueden observarse en el síndrome de Goltz y en el síndrome MIDAS, ambos de herencia ligada al cromosoma X y que afecta predominantemente a mujeres. El síndrome de Goltz se caracteriza por presentar áreas cutáneas de atrofia e hiperpigmentación de la piel, telangiectasia, depósitos localizados de grasa superficial, así como un pelo frágil y escaso en el cuero cabelludo. Este síndrome puede presentar alteraciones oculares; coloboma de iris y coriorretiniano, microftalmía y rara vez anoftalmía unilateral 31. En general no cursa con alteraciones neurológicas y las lesiones cutáneas son fáciles de diferenciar de las asociadas a la varicela fetal. El síndrome MIDAS es más raro, pero tiene una mayor trascendencia neurológica. MIDAS es un acrónimo que señala la existencia de microftalmía, aplasia dérmica y esclerocórnea. Las lesiones dérmicas (atrofia e hiperpigmentación) se localizan en la mitad superior del cuerpo, en particular en la cara y el cuello, siguiendo una distribución que recuerda las líneas de Blashko. Este síndrome puede presentar anomalías cerebrales como microcefalia, agenesia del cuerpo calloso y displasia cortical focal 66.
Lesiones tisulares subcutáneas hamartomatosas congénitas e hipertrofias corporales
Por hamartoma cutáneo congénito entendemos masas tumorales no neoplásicas que muestran una mezcla anormal de componentes habituales de los tejidos. El recién nacido puede presentar algunos síndromes hamartomatosos, algunos de ellos asociados a trastornos funcionales y estructurales del SNC.
El síndrome de Proteus es un síndrome hamartomatoso de expresión variable, caracterizado por gigantismo parcial, sobre todo de manos y pies, asimetría de las extremidades, hiperplasia cutánea plantar, nevos lineales verrugosos e intradérmicos, parches de shagreen y una combinación variable de tumores subcutáneos (linfangioma, hemangioma), así como macrocefalia e hiperostosis de los huesos de la bóveda craneal, de la cara y la mandíbula 67. Las lesiones cutáneas pueden seguir las líneas de Blaschko. Con escasa frecuencia están presentes malformaciones del SNC como: hemimegalencefalia, síndrome de Dandy-Walker, trastornos de la migración neuronal, hipoplasia del cuerpo calloso y atrofia cerebral. Se ha referido que alrededor de 20 % de estos niños presentan convulsiones, en ocasiones durante el período neonatal y un 20 % algún grado de trastorno cognitivo 67-69. Aunque el cuadro clínico se desarrolla después del período neonatal, se han descrito fetos y recién nacidos con signos característicos de este síndrome: hemihipertrofias llamativas, gigantismo de manos y pies, malformaciones vasculares, macrocefalia e hemimegalencefalia 70. Sin embargo, el diagnóstico suele ser tardío, cuando progresivamente se va acentuando la expresión durante la niñez y basarse el diagnóstico en hallazgos clínicos, radiológicos y evolutivos 67,68.
La lipomatosis encefalocraneocutánea es un raro síndrome neurocutáneo esporádico, que puede observarse desde el nacimiento y que se caracteriza por lipodermoides unilaterales que afectan a la conjuntiva, la esclera o los párpados, lipomas unilaterales sobre el cráneo, la cara y el cuello, y alteraciones cerebrales estructurales ipsolaterales. Por lo tanto, este es un trastorno que se limita a un lado del cráneo, la cara y el cerebro y los hallazgos característicos se han observado antenatalmente mediante ecografía fetal 71. Este cuadro puede cursar con convulsiones en la infancia y retraso mental. En los estudios de neuroimagen se observa atrofia cerebral, calcificaciones intracraneales y particularmente quistes porencefálicos 72,73. Es controvertido si este síndrome representa o no una variante del síndrome de Proteus y se ha sugerido que ambas entidades representan un continuum del mismo mosaicismo somático 73.
Diversos cuadros pueden cursar con hemihipertrofia. En el síndrome de Beckwith-Wiedemann 90 % de los pacientes presenta un nevo flamígero sobre la glabela y algunos presentan hemihipertrofia. Este síndrome cursa microcefalia leve y aproximadamente un 50 % de los pacientes presentarán trastorno cognitivo leve a moderado 31. El síndrome de Klippel-Trénaunay-Weber puede presentar un cuadro de gravedad variable caracterizado por hemangiomas subcutáneos, varicosidades, flebectasia y a veces fístulas arteriovenosas. La hemihipertrofia puede coincidir o no con las anomalías vasculares, gigantismo de manos y pies, sindactilia, polidactilia u oligodactilia. Ocasionalmente se observa un nevo flamígero de distribución similar a la observada en el síndrome de Sturge-Weber y estos pacientes presentarán trastorno cognitivo.
Piel arrugada y/o lipoatrofia generalizada
La presencia de una piel arrugada que recuerda a la de un viejo junto con marcada lipoatrofia generalizada en un recién nacido sugiere el síndrome progeroide neonatal o síndrome de Wiedemann-Rautenstrauch. Es una rara enfermedad autosómica recesiva en la que el recién nacido parece prematuramente envejecido y que se caracteriza por lipoatrofia generalizada, pero con almohadillas de grasa en la región lateral superior a las nalgas, piel arrugada e hipotricosis en cuero cabelludo, cejas y pestañas. Además, existe macrocefalia con venas prominentes en el cuero cabelludo (seudohidrocefalia), cara triangular, micrognatia y dientes neonatales. Esta enfermedad con frecuencia letal durante el primer año de vida, cursa con leve a moderado trastorno cognitivo en supervivientes 74. Es fácil de distinguir de otras que presentan lipoatrofia generalizada en el recién nacido como la lipodistrofia congénita de Berardinelli-Seip tipo 2 (BSLC2), la cual, a diferencia de la forma juvenil (BSCL1), cursa con retraso psicomotor o mental en la mayoría de los pacientes 75.
Una piel arrugada que afecta particularmete a las manos, los pies y el abdomen, se observa en el síndrome de la piel arrugada. Este asocia retraso del crecimiento intrauterino y posnatal, microcefalia y retraso mental. Se ha postulado que este y el síndrome de cutis laxa con retraso del crecimiento y del desarrollo representan distintas formas de presentación del mismo síndrome.
Piel rígida
La dermopatía restrictiva es una rara genodermatosis recesiva caracterizada por un crecimiento y diferenciación anormal de la piel que determina una piel delgada, rígida y adherente que limita el movimiento del feto y produce una secuencia deformativa por acinesia fetal: polihidramnios, facies dismórfica, contracturas articulares múltiples y muerte neonatal temprana por insuficiencia respiratoria. Otros hallazgos característicos son una boca pequeña y redonda, nariz pequeña, orejas de implantación baja y suturas craneales dehiscentes 76. El carácter marcadamente dismórfico del cuadro clínico hace que la naturaleza cutánea pueda pasar inadvertida. El diagnóstico diferencial se establece con enfermedades neuromusculares con grave secuencia deformativa por acinesia fetal, síndrome cerebrooculofacial-esqueleto (COFS), el síndrome de pterigium múltiple letal y en particular con el síndrome de Neu-Laxova 77.
En conclusión, numerosas entidades con riesgo neuroevolutivo cursan con alteraciones cutáneas como atrofia, cambios pigmentarios, anomalías vasculares y hamartomas o sobrecrecimientos. Reconocer estas alteraciones cutáneas es una primera y esencial etapa para el diagnóstico, el cual permitirá conocer los riesgos neuroevolutivos, establecer el seguimiento y en ocasiones iniciar intervenciones preventivas o terapéuticas precoces. La aproximación inicial al diagnóstico corresponde al neonatólogo, pero debido a la heterogeneidad genética y clínica, así como a la complejidad de la relación genotipo-fenotipo de las entidades que involucran a la piel y al SNC, el diagnóstico certero precisa en ocasiones de la ayuda de expertos en dermatología y en no pocas ocasiones de expertos en genética clínica.
