




La miocardiopatía restrictiva (MCR) es una entidad poco frecuente en pediatría (incidencia del 2,5-5% del total de miocardiopatías pediátricas)1. Se caracteriza por disfunción diastólica con función sistólica preservada y grosor miocárdico normal2. La mayoría de los casos son idiopáticos. Entre las causas conocidas destacan los casos secundarios a mutaciones genéticas (principalmente genes sarcoméricos) y a enfermedades infiltrativas (poco frecuentes en pediatría)3. Se han publicado casos de MCR pura o con fenotipos mixtos asociados a hipertrabeculación (MCNC) o hipertrofia4. El tratamiento médico no ha mejorado el pronóstico y existe rápida progresión de la hipertensión pulmonar (HTP) y riesgo de muerte súbita5. Por ello, se debe considerar el trasplante cardíaco (TC) de forma precoz.
Las variables se expresan como mediana (intervalo intercuartílico).
Se presenta un estudio retrospectivo de 9 pacientes con MCR diagnosticados en nuestro centro entre septiembre del 2002 y septiembre del 2019. La mediana de seguimiento fue 95,57 meses (0,76–209,74) y la de diagnóstico 24,02 meses (0,00-176,03). Las características de los pacientes se describen en la tabla 1. Todos presentaban una miocardiopatía primaria excepto 1/9 con sospecha de fibrosis endomiocárdica secundaria a esquistosomiasis6. En 6/9 el fenotipo fue MCR pura (mediana edad al diagnóstico 35,99 meses [1,81-176,03]) y en 3/9 fenotipo mixto de MCR/MCNC (mediana edad al diagnóstico 0,53 meses [0-123,30]); 2/9 tenían antecedentes familiares de miocardiopatía: 1/2 MCNC en familiar de primer grado y 1/2 MCR en familiar de primer y segundo grado. Se realizó estudio genético en 5/9: 1/5 negativo, 1/5 se diagnosticó de síndrome de Alstrom y 3/5 presentaban una mutación patogénica en genes sarcoméricos (1/3 en el gen de la troponina T [TNNT2] y 2/3 en el gen que codifica la cadena pesada de la betamiosina cardíaca [MYH7]); 6/9 pacientes iniciaron con clínica de insuficiencia cardíaca (IC): 4/6 con MCR pura y 2/3 con fenotipo mixto MCR/MCNC.
Características demográficas, electrocardiográficas, ecocardiográficas y hemodinámicas de los pacientes con miocardiopatía restrictiva
| Caso 1 | Caso 2 | Caso 3 | Caso 4 | Caso 5 | Caso 6 | Caso 7 | Caso 8 | Caso 9 | |
|---|---|---|---|---|---|---|---|---|---|
| Edad media (años) | 10,5 | 4,0 | 0,05 | 2,0 | 0,15 | 14,6 | 10,3 | 0,08 | 0,5 |
| Sexo | Mujer | Varón | Varón | Varón | Varón | Mujer | Mujer | Mujer | Varón |
| Tipo MCR | Secundaria Es | Primaria | Primaria | Primaria | Primaria | Primaria | Primaria | Primaria | Primaria |
| Fenotipo | MCR | MCR | MCR/MCNC | MCR | MCR | MCR | MCR/MCNC | MCR/MCNC | MCR |
| AF Miocar | No | No | No | No | Sí | No | No | No | Sí |
| Estudio genético | NR | NR | Neg | NR | Síndrome de Alstrom | Mut gen TNNT2 | NR | Mut gen MYH7 | Mut gen MYH7 |
| Síntomas IC | Sí | Sí | Sí | No | Sí | Sí | No | Sí | No |
| Electrocardiograma | |||||||||
| FC (lpm) | ND | 87 | 83 | 84 | 125 | 75 | 98 | 103 | 100 |
| PR(ms) | ND | 130 | 160 | 145 | 121 | 130 | 192 | 164 | 110 |
| Voltaje onda P (mV) | ND | 0,6 | 0,4 | 0,4 | 0,2 | 0,5 | 0,4 | 0,4 | 0,4 |
| QRS (ms) | ND | 89 | 90 | 90 | 65 | 125 | 90 | 82 | 98 |
| QRS (eje) | ND | 90 | 10 | 30 | 67 | 147 | 20 | 79 | 117 |
| Voltaje R en DII (mV) | ND | 1,0 | 0,8 | 1,1 | 0,5 | 0,5 | 0,5 | 1,2 | 0,7 |
| QTc | ND | 437 | 448 | 440 | 454 | 488 | 402 | 490 | 413 |
| Alteración repolarización | ND | InfradesST inferolat | Onda T Neg deriv laterales | Infrades ST y T Neg deriv inferolat | Onda T Neg deriv laterales | Onda T Neg deriv laterales | No | No | Onda T Neg deriv anterolat |
| Ratio P/QRS DII | ND | 0,36 | 0,5 | 0,36 | 0,4 | 1 | 0,8 | 0,33 | 0,57 |
| Arritmias | FibA | BAC | EV | No | No | TV/MS | No | No | No |
| Ecocardiografía | |||||||||
| AI (z-score) | 5,20 | 2,39 | 5,57 | 2,09 | 4,26 | 4,56 | 4,60 | 4,20 | 2,60 |
| IT (mmHg) | 100 | 35 | 80 | 54 | 58 | 48 | 66 | 90 | 15 |
| FEVI (%) | 55 | 55 | 70 | 54 | 60 | 69 | 59 | 50 | 73 |
| E (m/s) | ND | 0,40 | 0,83 | 0,37 | 1,23 | 1,07 | 0,80 | 0,80 | 1,50 |
| A (m/s) | ND | 0,58 | 0,32 | NR | 0,27 | 0,29 | 0,36 | 0,70 | 0,30 |
| E/A | ND | 0,69 | 2,60 | NR | 4,50 | 3,69 | 2,22 | 1,10 | 5,00 |
| E/e’ | ND | 7,60 | 8,30 | 7,40 | 13,70 | 21,40 | 8,50 | 16,80 | 10,00 |
| Cateterismo | |||||||||
| PAPm (mmHg) | 62 | 17 | 55 | 37 | NR | 37 | 23 | 58 | NR |
| PAI (mmHg) | 21 | 13 | 28 | 31 | NR | 25 | 21 | 26 | NR |
| PVItd (mmHg) | 24 | 15 | 27 | 21 | NR | 32 | 28 | 28 | NR |
| RVPi (UW·m2) | 23 | 2,2 | 12 | 6,1 | NR | 6,0 | 5,7 | 6,1 | NR |
| Estado actual | TCP/F | TC/Vi | TC/Vi | TC/Vi | LT/Vi | F | LT/Vi | TC/Vi | LT/Vi |
A: onda A flujo transmitral; AF Miocar: antecedente familiar de miocardiopatía; AI: aurícula izquierda; Anterolat: anterolaterales; BAC: bloqueo auriculoventricular; Deriv: derivaciones; E: onda E flujo transmitral; Es: esquistosomiasis; EV: extrasístoles ventriculares; F: fallecido; FEVI: fracción eyección ventrículo izquierdo; FibA: fibrilación auricular; Gen: genética; IC: insuficiencia cardíaca; Inferolat: inferolateral; Infrades: infradesnivelación; IT: insuficiencia tricuspídea; lpm: latidos por minuto; LT: libre de trasplante; MCR: miocardiopatía restrictiva; MCR/MCNC: miocardiopatía restrictiva mixta asociada a hipertrabeculación; ms: milisegundos; MS: muerte súbita; Mut: mutación; mV: milivoltios; ND: no documentado; Neg: negativo; NR: no realizado; PAI: presión aurícula izquierda; PAPm: presión arteria pulmonar media; PVItd: presión telediastólica del ventrículo izquierdo; Sdr: síndrome; QTc: intervalo QT corregido; RVPi: resistencias vasculares pulmonares indexadas; TC: trasplante cardíaco; TCP: trasplante cardiopulmonar; TV: taquicardia ventricular; Vi: vivo.
Todos los pacientes de los que se disponían datos del electrocardiograma (8/9) presentaron una ratio P/QRS elevada (0,5 mV [0,33-1,00]) a expensas del incremento de voltaje onda P (0,40 mV [0,20-0,60]) y disminución amplitud QRS (7,5 mV [5-12]). Durante el seguimiento, 4/9 pacientes desarrollaron arritmias (mediana de meses tras el diagnóstico): uno hemiparesia izquierda secundaria a episodio de fibrilación auricular (36,50), uno extrasístoles ventriculares frecuentes (9,30), otro bloqueo auriculoventricular (60,65) y otro taquicardia ventricular no sostenida con muerte súbita (0,76).
Ocho de 9 pacientes presentaban HTP estimada a través del gradiente ecocardiográfico de la insuficiencia tricuspídea (62mmHg [35-100]). Se realizó estudio hemodinámico en 7/9 pacientes (tabla 1).
Tras una mediana de 7,9 años de seguimiento, 3/9 pacientes estaban libres de muerte súbita o TC (fig. 1); 5/9 fueron trasplantados y 1/9 fue falleció súbitamente estando en lista de trasplante (caso 6); 4/5 pacientes trasplantados recibieron TC a los 61,37 meses del diagnóstico (39,72-141,04). El motivo del trasplante fue disfunción biventricular severa en 1/4 (caso 2), requiriendo soporte con oxigenación por membrana extracorpórea venoarterial y, posteriormente, asistencia biventricular previa al trasplante. En 3/4 (casos 3, 4 y 8) la indicación fue clínica de IC e HTP severa. El caso 3 presentaba presión pulmonar suprasistémica y RVPi 12UWm2, iniciándose asistencia ventricular izquierda para reducir resistencias pulmonares previa inclusión en lista de TC a las 4 semanas (RVPi 3,4UWm2), trasplantándose tras 11 meses de soporte circulatorio. El caso 1 procedía de otro centro y, al ser valorado, se objetivó HTP severa e irreversible (PAPm 62mmHg y RVPi 23UWm2), por lo que recibió trasplante cardiopulmonar. La supervivencia a los 4 años del trasplante fue del 100%. El caso 1 falleció a los 4,4 años del trasplante por rechazo pulmonar crónico.
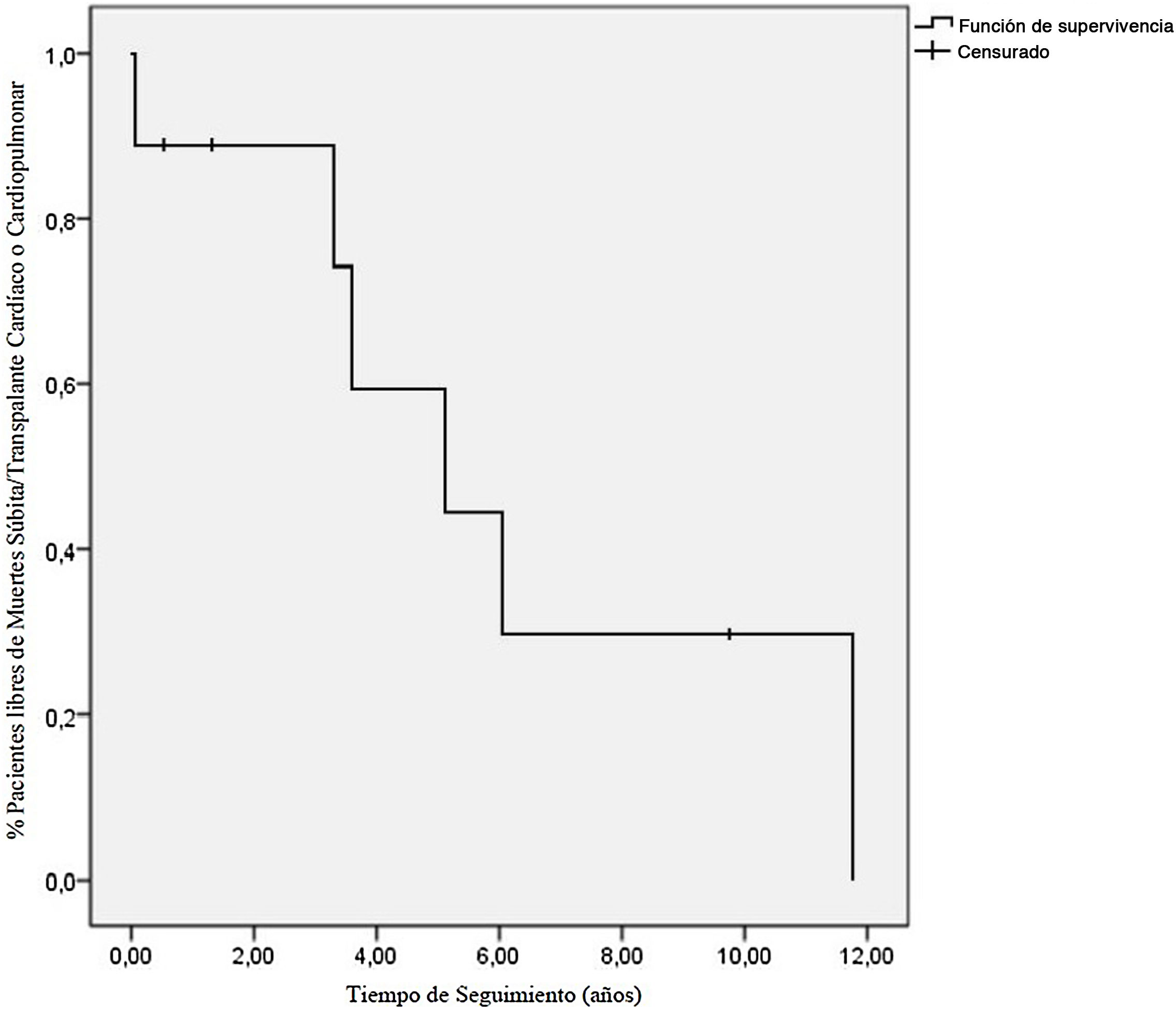
Como conclusiones, este artículo expone la serie española de pacientes pediátricos con MCR más extensa publicada, una entidad infrecuente en pediatría con una evolución clínica muy desfavorable y supervivencia libre de muerte o trasplante del 20% a los 5 años. La colocación de marcapasos o desfibrilador implantable en prevención primaria se debe valorar de forma precoz por el riesgo elevado del desarrollo de arritmias, incluyendo bloqueo auriculoventricular y muerte súbita1. La HTP es una complicación frecuente que puede contraindicar el TC, por lo que este se debe considerar de forma temprana tras la aparición de la HTP, así como por la mala evolución y la falta de tratamiento. En los pacientes con RVPi elevadas se puede contemplar la asistencia ventricular previa al trasplante. Debido a las complicaciones y al mal pronóstico de esta entidad, consideramos de especial importancia su derivación precoz para diagnóstico y seguimiento a centros de alta especialización con unidades de arritmias pediátricas, insuficiencia cardíaca y TC pediátrico.
